
ID research, is that all there is?
Behe published his book “Darwins Black Box” in 1996. In it he stated the principle of “irreducible complexity” and claimed that, amongst other things, the clotting system and the eubacterial flagella were irreducibly complex, and were not evolvable. Since that time, researchers have uncovered significant evidence for the evolution of both the clotting system and the eubacterial flagella, what has Behe been researching while this has been going on? Well at a recent ID conference Behe has apparently produced a calculation that shows that the evolution of new binding sites between proteins and things such as other protein, DNA and small molecules is so unlikely as to be impossible.
Now, I’m a pharmacologist, and my stock in trade is binding sites, so I was very interested in this. Binding sites are ubiquitous in biological systems; proteins bind together to form complexes, such as the respiratory complex in the mitochondrion (the cells power house), proteins bind to DNA in cell replication, and small protein hormones such as insulin bind to larger receptor proteins to cause various important cellular effects. So in effect, Behe is saying that an enormous part of the cell’s biology is vanishingly unlikely to have evolved. This is a very strong claim.
I’m very suspicious of this claim for several reasons. One is the phenomenon of “non-specific binding”, which is the tendency, when you are trying to follow the binding of a specific hormone to a specific receptor, for your specific hormone to bind to everything under the sun. That is, proteins tend to stick together by default, and it is not to hard to imagine this generalized binding evolving into a more specific binding. Also, a lot of experimental work has been done on evolving binding sites, especially by Jack Szostak, so it seems a very strange claim in the face of experimental evidence that binding sites can evolve. Unfortunately we don’t have Behe’s text from the Biola talk, but we do have one from a recent talk where he briefly discusses these issues.
I’ll use the text of this talk to illustrate some of the issues involved, as he apparently has kept the core concepts intact in the Biola conference talk. In the 2002 talk Behe uses the example of the Bovine Pancreatic Trypsin Inhibitor (BPTI). I’ll use the same protein to show how wrong he is. All quotes come from Behe’s 2002 talk.
Drawings of the bacterial flagellum picture proteins as bland spheres or ovals, but each protein in the cell is actually itself very complex. This ribbon drawing of bovine pancreatic (trypsin) inhibitor gives a little taste of that complexity. Now proteins are polymers of amino acid residues and some structural features of proteins require the participation of multiple residues. For example, up here, I know it’s hard to see, but this yellow link is called a disulfide bond.
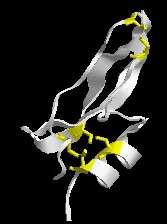
In biochemistry people tend to refer to amino acids as “residues”, so when Behe talks of residues he means amino acids. In the absence of Behe’s picture I’m using my own ribbon diagram of the BPTI. In ribbon diagrams the messy amino acid structure is idealized as ribbons and strings. The exception being the amino acid cysteine, which is here shown as yellow sticks. Two cysteines close together interact to form a disulfide bond. This disulfide bond is relative weak compared to the bonds that form the actual protein, but they are strong enough to help the protein keep its finger-like shape, rather than having the chains flop around aimlessly. In the case of BPTI, it has three disulfide bonds (i.e. 6 cysteines). Similar interactions can occur between other amino acids to help keep a protein in a particular shape, and these relatively weak interactions between amino acids are also involved in binding separate proteins together.
A disulfide bond requires two (cysteine) residues. Just one (cysteine) residue can’t form such a bond. Thus in order for a protein that did not have a disulfide bond to evolve one, several changes in the same gene have to occur. Thus in order for a protein that did not have a disulfide bond to evolve one, several changes in the same gene have to occur. Thus in a sense, the disulfide bond is irreducibly complex, although not really to the same degree of complexity as systems made of multiple proteins.
“Several changes”? Most proteins have at least one cysteine in them, all you would need is to have a second cysteine to be inserted near the first one. Here is a diagram of a mutant BPTI where two of the cysteines have been replaced. Note the overall similarity to the structure of the native BPTI. To get from this structure to BPTI we need exactly two mutations.
[img=l noborder width=157 height=224]http://www.pandasthumb.org/archives/images/BPTI_mut_s.jpg”/>
Thus in a sense, the disulfide bond is irreducibly complex, although not really to the same degree of complexity as systems made of multiple proteins. The problem of irreducibility, irreducibility in proteins is a general one. Whenever a protein interacts with another molecule as all proteins do, it does so through a binding site whose shape and chemical properties closely match the other molecule. Binding sites however, are composed of perhaps a dozen amino acid residues, and binding is generally lost if any of the positions are changed.
This is not true. One reason for this is that the amino acids that make up proteins come in general structural classes (acidic, basic, aromatic), so it is usually possible to substitute an amino acid of one structural class for another of the same structural class to get fairly similar function. Sometimes changing one amino acid will destroy binding, but most times reasonable binding remains. I’ll go into this shortly, first lets look at the interaction between BPTI and its binding site on trypsin..
[img=l noborder width=167 height=224] http://www.pandasthumb.org/archives/images/BPTI_trypsin_s.jpg “/>
Here I show the interaction between BPTI and bovine trypsin using a ribbon diagram. BPTI is the green ribbon (I’ve not shown the disulfide bonds for simplicity) and trypsin is the blue. Most people should have at least have heard of trypsin, it is an enzyme that helps us digest protein. There is a pocket in the structure of trypsin where the proteins it breaks down fit into, this is the binding site.
Part of BPTI is a loop that fits into this binding site on trypsin. You can see on the diagram where the loop fits in, partially obscured by the tyrpsin molecule itself. 7 amino acids in BPTI bind to amino acids in the pocket in trypsin, preventing it from binding to the proteins trypsin is supposed to break down (hence the name bovine pancreatic trypsin inhibitor). BPTI is part of family of proteins called Kunitz proteins. BPTI and similar proteins regulate the activity of enzymes like trypsin, other Kunitz proteins are toxins in snake venoms.
While 7 amino acids are involved in binding of BPTI to trypsin, the key determinant of BPTI binding is the amino acid at the very tip of the loop (called the P1 site) that penetrates into the heart of the binding pocket of trypsin. In my diagram I’ve shown in yellow an amino acid that occurs at this P1 site, but I’ve omitted the remainder of the amino acids on the loop that bind to trypsin for clarity.
The P1 site is responsible for over 50% of the interaction between the BPTI and tryspin. Normally this is the basic amino acid lysine. Substituting 10 different amino acids at this position resulted in a spectrum of binding (and trypsin inhibition) from weak to very strong (1). Basic arginine could substitute for basic lysine with little change in affinity. Even substituting an acid amino acid for the basic lysine still resulted in reasonable binding. There are two key messages here. One is that is that you do not need this critical residue to be a specific amino acid to get binding or functionality. The second is that binding is not an all or nothing affair, and that we get a graded change in binding with changing amino acids. Thus Behe’s claim “binding is generally lost if any of the positions are changed” is shown to be incorrect for his own example. A lysine to valine mutation at P1 converts the trypsin inhibitor into an inhibitor of human neutrophil elastase, so you can get new functionality quite easily without changing each and every amino acid in the binding loop.
Now, you might object that this is just one mutation, when all the others are in place, so Behe’s suggestion still has some validity. Remember that the P1 position accounts for over 50% of the interaction, so it is the key amino acid. Also, in another paper(2) they mutated 4 of the 7 amino acids, and were able to convert a trypsin specific BPTI to a porcine pancreatic elastase (PPE) inhibitor in single mutational steps. The wild type PBTI had no affinity for PPE, and went to a reasonably high affinity binding with just one mutation, which was refined to very high affinity binding with subsequent mutations. Again, using Behe’s own example, we can show his proposition is false.
One can then ask the question, how long would it take for two proteins that originally interact to evolve the ability to bind each other by random mutation and natural selection, if binding only occurs when all positions have the correct residue in place. Emphasis added by IFM
As we have seen, his argument comes apart here. Binding does not occur only with all residues in place. Let’s have a look at another BPTI, example (3). It has three disulfide bonds as we saw above. However, even versions with only one disulfide bond will bind and inhibit trypsin. Four of all 15 possible single disulfide variants bound reasonably well, including one non-native variant. What’s more, if the disulphide bonds were broken, the proteins still bound reasonable well (3). Thus, even with major structural changes, you can still get reasonable binding of the peptide to trypsin.
Although it would be difficult to experimentally investigate the question, the process can be simulated on a computer. . . .(mentions equation that does this, but gives no details IFM).. The yellow dot is the time expected to generate a new disulfide bond in a protein that did not have one if the population size is 100,000,000 organisms. The expected time is roughly a million generations. The red dot shows that the expected time needed to generate a new protein binding site would be 100 million generations.
As we have seen, it is not that difficult to experimentally investigate the question in Behe’s example, BPTI. There new functionality and binding was generated rapidly. Jack Szostak (4) has been evolving binding sites de-novo for some time now, and no such 100 million generation limit is apparent. Also, a recent mutation in humans has resulted in a new disulfide bond binding site in the protein apolipoprotein A, which provides increased functionality to the mutant protein.
Behe’s argument at Biola was essentially similar to the one given here, so Behe has spent over 6 years developing an equation that gives results that a simple literature search would have show are completely wrong.
This is a general problem for ID apologists, as many including Dembski have claimed that protein structures are inflexible and do not change readily, while in fact there is a wealth of research showing that protein function is very flexible, and that novel functions are rather readily evolveable(5). The recent claim that “..[over the past 10 years]intelligent design has laid the foundations for a general biology whose fundamental organizing principle is intelligent agency and not blind natural forces” rings hollow when the chief proponents aren’t even familiar with the enormous body research that contradicts their basic claims, and even for their chosen examples.
(1) Helland R, Otlewski J, Sundheim O, Dadlez M, Smalas AO. The crystal structures of the complexes between bovine beta-trypsin and ten P1 variants of BPTI. J Mol Biol. 1999 287:923-42.
(2) Kiczak L, Kasztura M, Koscielska-Kasprzak K, Dadlez M, Otlewski J. Selection of potent chymotrypsin and elastase inhibitors from M13 phage library of basic pancreatic trypsin inhibitor (BPTI) Biochim Biophys Acta. 2001 1550:153-63.
(3) Krokoszynska I, Dadlez M, Otlewski J. Structure of single-disulfide variants of bovine pancreatic trypsin inhibitor (BPTI) as probed by their binding to bovine beta-trypsin. J Mol Biol. 1998 275:503-13.
(4) Huang Z, Szostak JW. Evolution of aptamers with a new specificity and new secondary structures from an ATP aptamer. RNA. 2003 9:1456-63.
(5) Gerlt JA, and Babbitt PC. Mechanistically diverse enzyme superfamilies: the importance of chemistry in the evolution of catalysis. Curr Opin Chem Biol 1998. 2:607-12.