
An Open Letter to Dr. Michael Behe
Dear Dr. Behe
I have recently read your response to Abbie Smith’s article on the HIV-1 protein VPU. Ms Smith showed how Vpu’s recently evolved viroporin activity directly contradicts your statement that HIV has evolved no new biding sites since it entered humans (Edge of Evolution, page 143 and figure 7.4, page 144 ). I was greatly disappointed in your response. I must admit to having a special involvement in this case. Firstly, I drew the illustrations for Ms Smith’s article, and its follow up. But secondly, as a member of my professional association’s education committee, I am directly concerned with the support and nurturing of the new generations of enquiring minds, those that we will pass the torch of enquiry on to when we retire. It is in this regard that your response very disturbing. It is almost the exact opposite of what a concerned scientist and science communicator should have done.
It was bad enough that you chose to ignore her for over two months and then did not do her the courtesy of replying on her blog (1). It was bad enough that you chose to start by belittling her and playing the “I’m a Professor and she is a mere student” card (conveniently ignoring the fact that she actually works on HIV). This is particularly egregious in science, where we pay attention to the evidence and logic of an argument, rather than the letters after an author’s name. Doubly so if we wish to guide young scientists into a demanding profession.
But by far the worst, you ignored her core argument. That in the space of a decade HIV-1 Vpu developed a series of binding sites that made it a viroporin, a multisubunit structure with a function previously absent from HIV-1. Dr. Behe, it is not enough to cite a generalist review and claim that the differences between HIV-1 strains are “not all that great”. You actually have to show why Vpu developing binding sites to form a multi-subunit structure with a novel function does not falsify your claim that HIV has developed no new binding sites. Ironically, the very paper you cite to dismiss Ms Smith contains evidence of at least two new binding sites in HIV. I will not dwell on this any further, as Ms Smith is producing her own response.
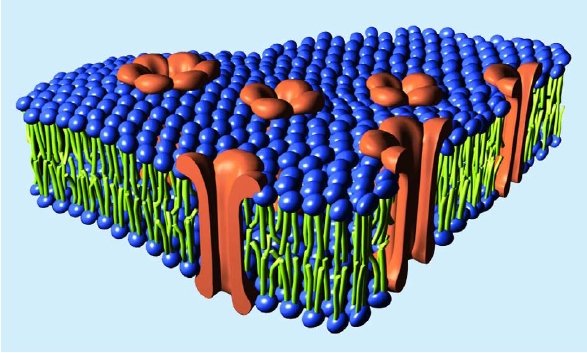
But I will comment on one other aspect of your response. Not content with dismissing Ms Smith, you make an incorrect statement as a central part of your argument.
Another, more important point to note is that I’m considering just cellular proteins binding to other cellular proteins, not to foreign proteins. Foreign proteins injected into a cell by an invading virus or bacterium make up a different category. [emphasis added here[by MB]] The foreign proteins of pathogens almost always are intended to cripple a cell in any way possible. Since there are so many more ways to break a machine than to improve it, this is the kind of task at which Darwinism excels. Like throwing a wad of chewing gum into a finely tuned machine, it’s relatively easy to clog a system — much easier than making the system in the first place. Destructive protein-protein binding is much easier to achieve by chance.
This is simply not true, either generally (2) or in the particular case of Vpu. Importantly, your statement shows that you do not understand what Vpu does. Vpu down regulates the surface protein CD4 (the Viroporin activity is something separate related to viral release). It does not “gum-up” CD4, it specifically binds to it, then binds to a separate protein (the βTrCP subunit of the SCFβTrCP ubiquitin ligase complex) in the Golgi apparatus (all except the C strains, which target the plasma membrane). This multiprotein complex links to the ubiquitin-proteasome pathway, where CD4 is ignominiously broken down by the cells own mechanisms. This CD4–Vpu–βTrCP complex is NOT mere “gumming up of the works” but a precise targeting of CD4 the proteosome by Vpu. This is not obscure, but has been well known for some time.
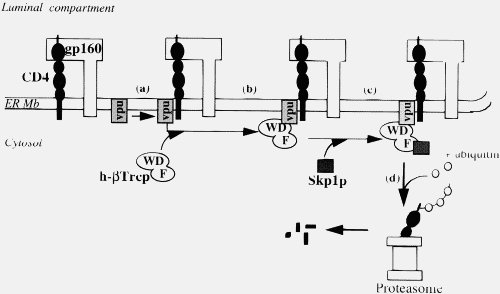
In the context of Ms Smith’s argument, HIV-1 Vpu has a new binding site, YRKL in the cytoplasmic alpha helical section, not present in SIVcpz Vpu, which efficiently targets Vpu to the Gogli complex, making the degradation process more efficient. Can you please explain why the appearance of a new targeting motif in HIV-1 Vpu is NOT an example of a new binding site.
The fact that you chose to dismiss Ms Smith on the basis of your ignorance of how Vpu actually works, as well as ignoring her central argument is of great concern. As scientists, we have a duty both to deal with arguments carefully, and to nurture upcoming young researchers. Dr. Behe, you have failed both these duties. You owe Ms Smith a complete apology for such behaviour.
Your sincerely, Ian Musgrave
(1) Unfortunately, I can’t reply on the Amazon blog, as Amazon does not remember my purchases there, perhaps some kind soul will repost my letter there.
(2) As a rule neuroscientists and molecular biologists get excited over toxins because they are so specific, not because they general gunge up things. However, this is not the place to discuss injectable toxins.
References:
Margottin F, et al., A novel human WD protein, h-beta TrCp, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Mol Cell. 1998 Mar;1(4):565-74.
Bell CM, et al., Molecular Characterization of the HIV Type 1 Subtype C Accessory Genes vif, vpr, and vpu. AIDS Research and Human Retroviruses. 2007, 23(2): 322-330.
Musgrave IF, Seifert R, Schultz G. Maitotoxin activates cation channels distinct from the receptor-activated non-selective cation channels of HL-60 cells. Biochem J. 1994 Jul 15;301 ( Pt 2):437-41.