
An Open Letter to Dr. Michael Behe (Part 2)
Dear Dr. Behe
Abbie Smith has recently responded to your reply to her article on the HIV-1 protein Vpu. To refresh your memory, Ms Smith showed that the recently evolved viroporin activity of HIV-1 Vpu directly contradicts your statement that HIV has evolved no new binding sites since it entered humans (see “Edge of Evolution”, page 145 and 146). I see you intend to reply to my open letter at your Amazon blog, rather than engaging in open discussion here, or better yet, doing Ms Smith the courtesy of replying on her own blog. I hope that at least this time you will reply to the key argument Ms Smith made:
HIV-1 M Vpu is a viroporin.
SIV Vpu is not a viroporin, HIV-1 O Vpu is not a viroporin. This is a new activity that evolved in HIV after the split from SIV over a 10 year timeframe and is part of the reason that the HIV-1 M clade is the most common type of HIV in the world.
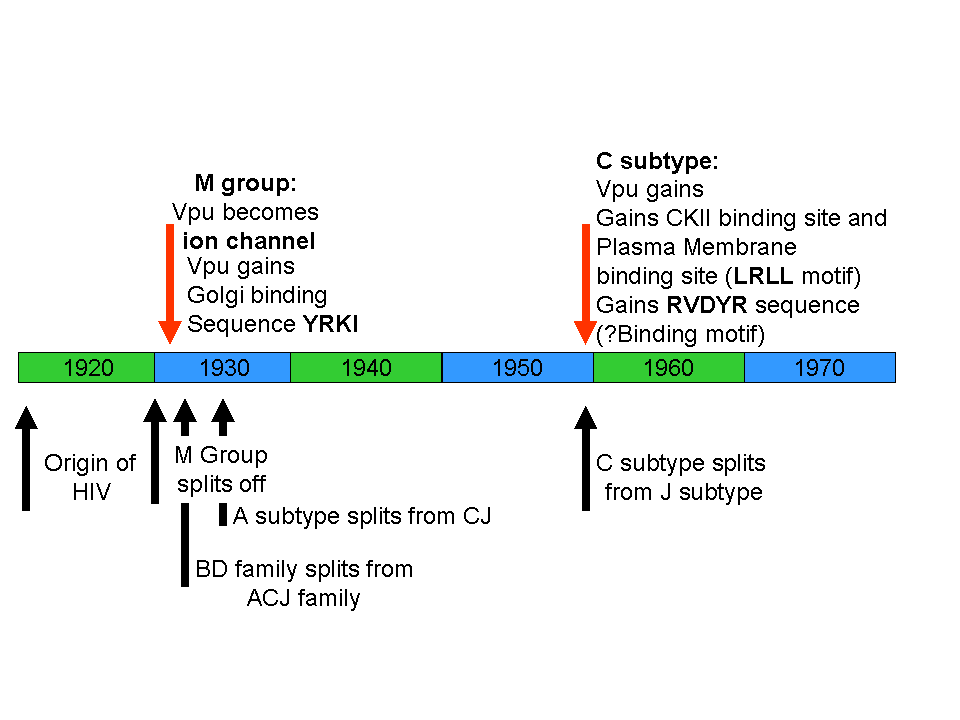 Timeline of the evolution of new binding sites in HIV. Note that several new binding sites develop after the evolution of HIV.
Timeline of the evolution of new binding sites in HIV. Note that several new binding sites develop after the evolution of HIV.
Just to remind you Dr. Behe, a viroporin is an ion channel, in the case of the HIV-1 Vpu, it is a gated cation channel, which appears relatively Na+ specific (Ewart et al., 1996, Bell et al., 2007, Menhert et al., 2007). To go from its original form to the multisubunit structure with a new function required the development of a new binding site, which involves more than a single amino acid substitution (Paul et al., 1998). Not just any binding site will do, or you get a higgledy-piggledy mass of agglomerated protein, not an ion channel with ion selectivity. Furthermore, this is a gated ion channel, not just a hole punched in the membrane, with a specific amino acid responsible for the gating (Mehnert et al., 2007).
Importantly, this mutation, producing a mini-“molecular machine”, is beneficial to the virus; it increases viral particle release, spreading HIV more efficiently (Paul et al., 1998). This is exactly what you demand in “Edge of Evolution”, viral protein-protein binding sites that are beneficial (see pages 139, 145 and 146). Incidentally, when on page 139 you state that there are no new reports of viral protein-protein binding, your citation to support this statement (21, Wang 2002), is a paper on how viruses recognize receptors, which is entirely irrelevant to your claim, this paper does not address surveys of HIV for new protein-protein interactions at all [1].
Dr. Behe, you can’t get out of this by claiming that the binding site is too simple, or not between completely different proteins. After all, your example binding site for humans is the Haemoglobin S point mutation. This is a one amino acid mutation that just destabilises the protein (what’s wrong with the Apolipoprotein A1 mutation that confers resistance to heart attacks?). If a one amino acid mutation that just causes haemoglobin to gunge together counts, then a multi-mutation event that leads to the formation of an elegant gated ion-channel which increases viral infectivity must count by your very own criteria.
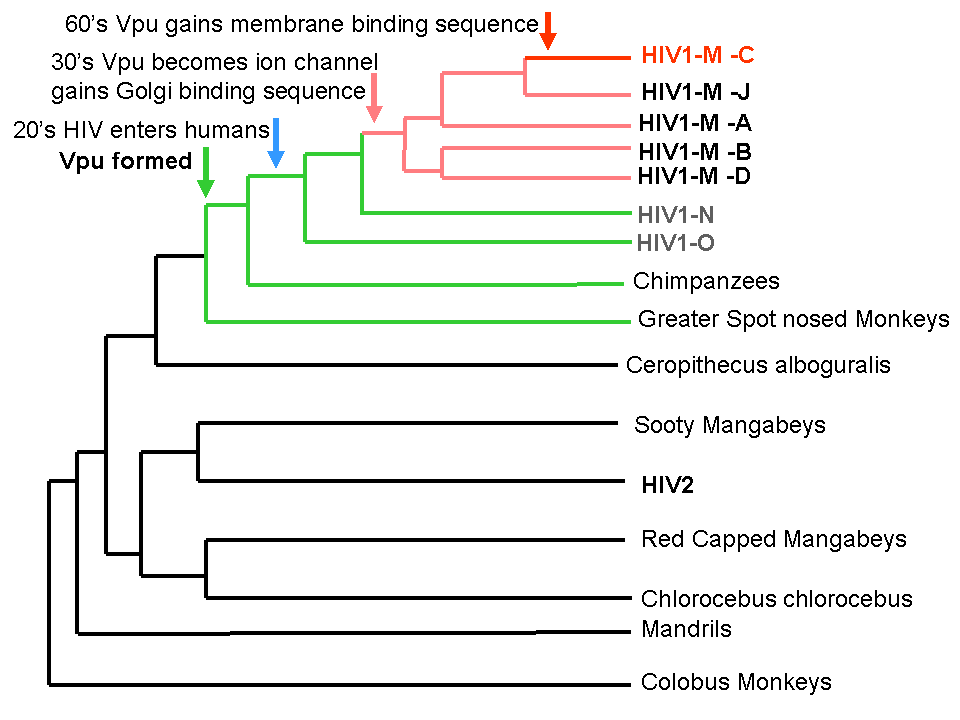 Evolution of HIIV binding sites in context of HIV binding.
Evolution of HIIV binding sites in context of HIV binding.
So, contrary to your claims in “Edge of Evolution” there is at least one protein that fulfils all your criteria as enumerated in that book, new viral-protein-protein interactions which forms new basic machinery that has a survival benefit for the virus. This evolved in a mere 10 years, with the notable absence of any Soros uplift fleet. Also in this time a new Golgi targeting sequence, YRKI evolved (Pacyniak et al., 2005). Again, this is a multisubunit binding site that provides a benefit for the virus by targeting the Vpu-CD4 complex to the Golgi apparatus. Then there is the recently evolved D/GXLRLL sequence in HIV-M subtype C which binds adaptor protein complexes on the cell surface, resulting in targeting of Vpu to the cell surface and more efficient viral spread (Hill et al., 2007). You dismiss these examples generically because you claim that
Like throwing a wad of chewing gum into a finely tuned machine, it’s relatively easy to clog a system
As I’ve shown previously, this is nonsense. We are talking about a coordinated binding to a specific acceptor site. Nor should there be a blanket ban of these types of protein binding sites anyway, a binding site is a binding site, you still need to develop the same specific holes and knobs, and electrostatic charges to get specific, high affinity binding, whether the binding sites results in the formation of a molecular machine or a receptor blocker. The development of the binding sites uses identical processes no matter what the downstream outcome. Again, since your own example is the single point mutation in haemoglobin, which just leads to it gluging up, you claim that a specific multisite mutation which targets a multimeric complex to the specific, defined cellular site is not a valid binding site rings rather hollow.
What did you expect a virus would do anyway? HIV-1 is a cut down parasite with 9 genes (HIV-2 has 10, there is a new gene in HIV-2 not present in SIV cpz due to duplication and subsequent mutation of the Vpr gene, and has novel binding targets and functions that Vpr does not. This invalidates your statement on page 139 that “No gene duplication has occurred leading to a new function”). The entire reason for most of these proteins is to bind to cellular proteins (that’s why parasites such as viruses are so dramatically simplified, they trick the bodies own cellular machinery. By defacto ruling out of court viral protein-cellular protein interactions for reasons that don’t make any sense in the light of your own arguments and examples, you have ruled out 99% of the potential evolution of these viral proteins.
But even here you are inconsistent; on page 139 you lament that the HIV virus has not developed any new cell surface binding sites. This is a viral-protein-cellular protein binding site, but it seems to count when you can’t find an example. But unfortunately you missed the development of CXCR4 coreceptor binding. This can happen within the course of a single infection. The viral receptor binding protein gp120 mutates and switches from binding the CCR5 coreceptor to the CXCR4 coreceptor (Salemi et al., 2007). All in the duration of one infection, again with the absence of any Soros uplift fleet. Once more your references fail you Dr. Behe, you cite reference 19 (Demma et al., 2006) as evidence that HIV has not developed the ability to bind to other receptors and enter new cellular hosts. Yet Demma et al., is not about HIV, it is about how mutations in SIV proteins such as env allow them to bind to new targets, allowing them to bind to receptors on new species cells. This is almost exactly the opposite of what you claimed. Of course, you dismiss this as not an example of a major biochemical novelty, but that is irrelevant. We are talking about proteins sticking together; your claim is that they can’t easily develop this capacity, irrespective of what the functional consequences are (what major biochemical novelty occurs in chloroquine resistance?). Again, this line of argument is completely blown away by your own haemoglobin S example.
To conclude, contrary to you claims that zero binding sites have developed in HIV, we can point to several, in Vpu alone that conform to your own criteria. When Ms Smith pointed this out to you, you ignored her core argument and replied with misdirection and misinformation. I sincerely hope that this time around you will fairly represent her arguments. Comparing someone to the cast of “Mean Girls” is annoying, but by completely avoiding the core argument, and dragging in nonsequiters is the worst disrespect of all.
However, this form of response, where you write on a blog I can’t post too, and I write on a blog you won’t post too, is very limiting. I hereby invite you to debate on a neutral site where there will be minimal distraction. I have created the “The VPU debate” weblog site, and I invite you to be a co-registrant. There we can concentrate on the mutations of Vpu and how they relate to your claims. I look forward to seeing you there.

Yours sincerely
A featherless biped named Ian Musgrave[2,3]
[1] This is a feature of a number of the references you cite, they are either irrelevant to your point, or don’t actually claim what you say they claim. Looking at your reference 21 (Wang 2002) again, this is supposed to support the claim that no new viral binding proteins have developed, but it is actually about determinants of viral surface binding. Your commentary on this paper doesn’t relate it to the claim it is supposed to support, but claims that viral protein binding only has to be weak and…well its not clear what you are trying to get at, here (certainly not the claim that there are now new HIV binding sites), probably that because only weak binding is needed you can find binding sites easily. Unfortunately for you, the HIV affinity for CD4 is 10 nM, what biologists consider high affinity binding, which makes your argument irrelevant.
\[2\] Strictly speaking, I'm a senior lecturer, not a professor. Although the Australia senior lecturer classification is roughly equivalent to a US professor position, the real professors here would get annoyed if I get an undeserved professor designation.
[3] Boring disclaimer: The University of Adelaide and I have a deal, they don’t speak for me, I don’t speak for them, so my views are mine alone, not official University views (like I would have that power, yeah)
References:
Bell CM, et al., Molecular Characterization of the HIV Type 1 Subtype C Accessory Genes vif, vpr, and vpu. AIDS Research and Human Retroviruses. 2007, 23(2): 322-330.
Demma LJ, et al., (2005) [ SIVsm quasispecies adaptation to a new simian host](http://pathogens.plosjournals.org/perlserv/?request=get-document&doi=10.1371/journal.ppat.0010003). PLoS Pathog. Sep;1(1):e3. Epub 2005 Sep 30.
Ewart GD, Sutherland T, Gage PW, Cox GB (1996) The Vpu protein of human immunodeficiency virus type 1 forms cation-selective ion channels. J Virol 70: 7108–7115.
Hill SM, et al., (2007) Modulation of the severe CD4+ T-cell loss caused by a pathogenic simian–human immunodeficiency virus by replacement of the subtype B vpu with the vpu from a subtype C HIV-1 clinical isolate. Virology, In press.
Goujon, C et al., (2007) [ SIVSM/HIV-2 Vpx proteins promote retroviral escape from a proteasome-dependent restriction pathway present in human dendritic cells ](http://www.retrovirology.com/content/4/1/2) Retrovirology, 4:2
Mehnert T, et al., Biophysical characterization of Vpu from HIV-1 suggests a channel-pore dualism. Proteins. 2007 Oct 1; doi: 10.1002/prot.21642.
Paul et al. (1998) Mutational Analysis of the Human Immunodeficiency Virus Type 1 Vpu Transmembrane Domain That Promotes the Enhanced Release of Virus-Like Particles from the Plasma Membrane of Mammalian Cells. J Virol, 72 (2): 1270.
Pacyniak E, et al., (2005) Identification of a region within the cytoplasmic domain of the subtype B Vpu protein of human immunodeficiency virus type 1 (HIV-1) that is responsible for retention in the Golgi complex and its absence in the Vpu protein from a subtype C HIV-1. AIDS Res Hum Retroviruses. May;21(5):379-94.
Salemi M, et al., (2007) [ Phylodynamics of HIV-1 in Lymphoid and Non-Lymphoid Tissues Reveals a Central Role for the Thymus in Emergence of CXCR4-Using Quasispecies. ](http://www.plosone.org/article/info:doi/10.1371/journal.pone.0000950) PLoS ONE. Sep 26;2(9):e950.
Wang J. (2002) Protein recognition by cell surface receptors: physiological receptors versus virus interactions. Trends Biochem Sci. Mar;27(3):122-6.