
An Open Letter to Dr. Michael Behe (Part 7)
Dear. Dr. Behe
I am pleased that you have acknowledged Vpu viroporin represents a real, de novo binding site.
Now if you had engaged with this in your response to Ms Smith, my respect for you would have risen immeasurably. To those of you not familiar with graduate and post graduate education, we actually want graduate students to disagree with us, robustly. After all, they are the ones carrying the torch of critical enquiry when we are gone. We don’t want them to accept our say so, “just because”. As scientists and educators our brief goes beyond just those PhD students we supervise, but to all engaged in critical enquiry, regardless of how we feel about their actual mode of delivery [1].
By “playing the man”[2] Dr. Behe, rather than engaging with Ms Smith’s arguments, you abrogated our responsibility as mentors and educators. Imagine the difference if you had dealt with Vpu Viroporin straight up. How about apologizing to Ms Smith now?
I do hope that you will now publish an erratum for your book, where you acknowledge Vpu viroporin. But again you engage in the “unimpressed” rhetoric. It matters not whether we are impressed or not by the outcome of the binding, the fact is that you have claimed that binding of two (or more) proteins to each other is statistically unlikely. It doesn’t matter what they do when bound (after all, as I have repeatedly pointed out, your own example is the haemoglobin S point mutation, which just glugs things up).
Your claim is that protein-protein binding can’t even get started, yet the type of binding that Vpu displays is exactly the type you need to build a proteosome, or the flagellum, which you are impressed by. What you claim is impossible in principle can be seen to actually happen in practice.
The Vpu viroporin is a gated ion channel, a mini-“molecular machine”; I know you don’t seem to be impressed by this, apparently because it doesn’t utilize ATP. But I divide my time equally between G-protein coupled receptors and signal transduction systems and ion channels, and you are ignoring the fact that ion channels, such as the Vpu ion channel, are major drivers of cellular activity. Changing ion gradients are critical for things from renal excretion to nerve activity. Everything from nicotinic receptors to renal sodium channels are built on the basic plan of Vpu.
I know you are impressed by large complexes such as the flagellum and the protesome, but biology doesn’t run on what you are impressed by. You dismiss a wide range of newly developed binding sites in HIV, such as the casein kinase binding site. Yet this is the canonical example of a regulatory binding site that links together important signal transduction cascades, everything from the binding of G-proteins to G-protein coupled receptors, to the protein kinase C activator cascades, to the MAPKinase regulatory cascades, to the calmoduin kinase binding that underlies nerve activity all rely on the same type of binding sites that you dismiss as “too simple”.
Dr. Musgrave misunderstands my point. I did not say that Vpu acted as a nonspecific wad of chewing gum.
And yes, you did, say the injected viral protein act as a nonspecific wad of chewing gum, its here in your Amazon blog and is quoted from chapter 8 of “Edge of Evolution”.
Like throwing a wad of chewing gum into a finely tuned machine, it’s relatively easy to clog a system — much easier than making the system in the first place. Destructive protein-protein binding is much easier to achieve by chance.
Now, Vpu doesn’t do that, it forms an elegant targeting system that places CD4 into the proteosome queue.
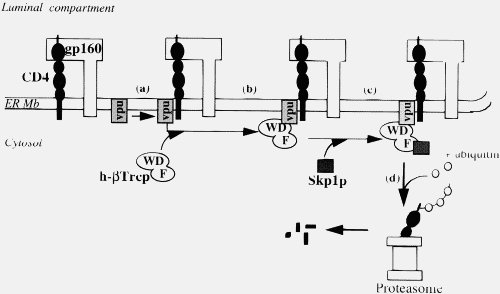
Your explanation of the “wad of chewing gum” effect in your most recent post is very different from what you wrote in you book and in your initial post. And while this version is more sophisticated (why didn’t you use that in your book), it is both biologically unrealistic and basically irrelevant. Your major claim is that binding sites need to develop two or more amino acid attachments simultaneously for any selectable binding. You spend ages on the mathematics of this. The probability of thee simultaneous amino acids mutations are the same whether it is a newly injected protein in a new host of opportunity or an established cellular protein.
The biological unreality is this:
.. initially, when during the course of evolution a viral or bacterial protein is injected for the first time into a cell, it is encountering a new environment, one it hasn’t adapted to before, and which hasn’t adapted to it.
Of course, in an established viral infection, such as HIV, this is not true. But more importantly, injected viral proteins act under exactly the same constraints as cellular proteins. A virus’s role is NOT to kill cells (although some viruses may do this in the course of an infection, they do it in a coordinated way). A virus is a parasite, its job is to get into the cell, take over the cellular machinery of the host and make many copies of itself and pass those copies on (some viruses release viral particles by coordinated cell lysis, but HIV buds off). It cannot afford its entry and replication machinery and budding machinery to bind to proteins willy-nilly, just like cellular proteins. It needs a coordinated response, indeed the Vpu binding to CD4 and attaching it to the proteosome is such a coordinated response, not willy-nilly jamming things up. If Vpu started out binding to any old protein, the viruses would be stuffed for sure. Lets just repeat that, viral proteins are under the same constraints as cellular proteins.
So, you argument is still irrelevant. But it is ironic that you are using a natural selection argument to why new binding sites cannot be found in existing cells. Pointing out that there are few new niches for new molecular machines hardly constitutes an “Edge of Evolution”.
In summary, the HIV virus has evolved several binding sites since it first infected humans. The binding sites are representative of a broad range of binding sites that are of key importance in cellular function. They evolved in a short timespan when virus effective population was orders of magnitude below the 1020 claimed necessary for binding site evolution. One of these binding sites makes a novel structure, a viroporin, which certainly out performs the HbS mutation. And that is just VPu, there is also a host of other binding sites which have only just been investigated (TRAIL upregulation CXCR4 binding, LTR binding etc.)
Thus Dr. Behe, you owe people an erratum for your book, and an apology to Ms Smith.
Yours sincerely
A male featherless biped named Ian Musgrave
[1] Maybe its because I’m Australian, we are a bit more “robust” than Europeans (having been mentored in
Prof Mike Rand’s department, Prof Rand was, ahhh, rather direct to all and sundry, the high and mighty got no mercy from him. You can spend time with my PhD student, The Heavy Metal Kid, for a while if you like, if you want “robust”.
[2] Australian Rules football jargon, it means attacking the player, rather than playing the game and capturing the ball. It is frowned upon severely (And yes, they still use “man” rather than person or some gender neutral term, it is Australian AFL after all)
(UPDATED: I’ve added some more quoted text to make things clearer)