
Structural phylogenetics and Kitzmiller vs. Dover
![[Figure 1 of Puente-Lelievre, Matzke et al. 2023, Tertiary-interaction characters enable fast, model-based structural phylogenetics beyond the twilight zone, bioRxiv]](/uploads/2024/CPL_etal_2023_Fig6_PDB_trees.png)
A slightly late Merry Kitzmas to all! The 2005 Kitzmiller v. Dover Area School Board case, where “Intelligent Design” went on trial (Matzke 2006) is now 18 years old. Amazingly, 18 years is the same number of years as between the 1987 Edwards v. Aguillard Supreme Court decision and Kitzmiller. I would say that the prominence of antievolutionism has receded in the latter 18 years, compared to numerous other culture wars, real wars, and challenges to the fundamentals of science-informed liberal democracy that have dominated the news in recent years.
In that sense, Kitzmiller has definitely had a positive impact. However, we should keep in mind that Alabama still has a warning label in its textbooks (see this story by Trisha Crain, 2023, al.com), several “critical analysis” antievolution policies remain on the books (Matzke 2015), for example the Louisiana Science Education Act of 2008 (NCSE 2023), and keeps evolving (Matzke 2015), Creation-museum-boosting young-earth creationist Mike Johnson is Speaker of the US House and third in line to the Presidency, the Texas Board of Education is still blocking textbooks with coverage of evolution and climate change that they don’t like, etc. As Rob Pennock always says, “stay vigilant!”
Longtime PT readers may recall some interesting scientific publications that came out of the Kitzmiller case, mostly reviewing the relevant science. Examples include the evolutionary origin of the immune system (Bottaro, Inlay, and Matzke 2006, Nature Immunology; see also posts at PT and NCSE), the evolutionary origin of complex adaptations (Scott and Matzke 2007, PNAS; PT post), or tracking the “evolution” of antievolution bills (Matzke 2016, Science; PT post; free online Supplemental Data). Another was on the evolution of the bacterial flagellum (Pallen & Matzke, 2006, Nature Reviews Microbiology; PT post), which I think crossed over into novel research on the origin of a complex adaptation, even though the article was mostly assembling previous findings into a coherent scenario. I have maintained an interest in the evolution of the flagellum, and I would like to present some new research that, in a roundabout way, derives from the flagellum work and from Kitzmiller.
I am part of a team on a Human Frontier Science Program research grant that is combining phylogenetic, modeling, and experimental approaches to further understanding the evolutionary origin of the bacterial flagellum. The group is headed by Matt Baker at the University of New South Wales (UNSW) in Sydney, Australia.
One challenge that has always been a barrier to using phylogenetics to understand the origins of the flagellum is the system’s great age. It may well be that the flagellum was present in the last common ancestor of (known, extant) bacteria, which probably means the system is 2.5+ billion years old. The other famous systems that the intelligent design (ID) movement latched onto were much younger: the eukaryotic cilium (or flagellum) is probably as old as eukaryotes, perhaps 1.5 billion years; the adaptive immune system and blood-clotting cascades arose within the vertebrates and so are less than 500 million years old. While phylogenetics is useful for understanding the origin of all these systems, even the bacterial flagellum, it’s just harder for the flagellum, as even the best conserved homologous proteins can have amino acid (AA) sequence similarity that has decayed to 30% or less. Homology detection, sequence alignment, and accurate phylogenies all become substantially harder as AA sequence similarity decays below 30%, especially below 25% and 20% and below. Eventually similarity decays to random background similarity and homology searches, alignments, etc. return nothing convincing, or, worse, they return something that is a little bit plausible but is actually just a chance match.
This zone below 30% AA sequence identity has long been known as the Twilight Zone of protein phylogenetics (this phrase goes back at least as far as Russell Doolittle in the 1980s, probably further). The main solution when homology/alignment/phylogenetics gets difficult due to loss of signal in the characters is: find more characters, ideally characters that are more slowly evolving. We already do this in many areas: if DNA sequence similarity is saturated, we can move to AA sequence, or morphological characters, both of which tend to be more slowly evolving, and thus preserve evidence of shared ancestry (shared derived characters) for longer.
Protein structure phylogenetics in the Twilight Zone
We do have another source of data in the Twilight Zone, which ought to be helpful: protein structure. It has long been known that protein structure is even more conserved than amino acid sequence. However, use of protein structure in phylogenetics has been limited by two things:
(1) Protein structures are very hard to experimentally characterize. Techniques like protein crystallography only work on certain proteins, and require a great deal of time, effort, and expensive equipment. Therefore, protein structures have historically only been available for a small fraction of the gene/protein sequences available after the DNA sequencing revolution.
(2) Converting a protein structure into a series of discrete characters that can be analyzed by phylogenetics software is a nontrivial problem.
For a long time, my best idea to solve problem #2 was to kidnap a few crystallographers and other protein structure experts and lock them in a basement until they came up with a manual character coding scheme for protein structures, and then used it to produce a character matrix. Much like paleontologists and morphologists manually describe the anatomy of their specimens and code this into a morphological character matrix for computational analysis, we could do the same thing with protein structure – at least if we can run faster than crystallographers.
However, two recent developments have radically changed what seems possible. First, AlphaFold2 and similar projects appear to have finally cracked the problem of predicting protein structure from AA sequence. While it’s not perfect, it appears to be pretty good. In 2024 (unlike 2020), we can input an AA sequence and get a pretty good structural prediction out.
Second, bioinformaticians (namely Martin Steinegger and colleagues) have figured out a way to code some of the 3-dimensional structural features of a protein structure into a linear alphabet of characters. This was done in the program Foldseek, used for homology searches deep into the Twilight Zone. The alphabet is called “3Di” (3-dimension).
At the University of Auckland, my postdoc Caroline Puente-Lelievre, and students and colleagues, were discussing and working with Foldseek, and it very gradually dawned on us that if the Foldseek 3Di alphabet was useful for homology search, it might be useful for phylogenetic inference as well.
We first explored this possibility on flagellum proteins, but then we realized that we had stumbled on something that might be the basis for improved phylogenetic inference for almost any protein clade that had diverged into the Twilight Zone. So we wrote a paper:
Puente-Lelievre, Caroline; Malik, Ashar J.; Douglas, Jordan; Ascher, David; Baker, Matthew; Allison, Jane; Poole, Anthony; Lundin, Daniel; Fullmer, Matthew; Bouckert, Remco; Kim, Hyunbin; Steinegger, Martin; Matzke, Nicholas J. (2023). Tertiary-interaction characters enable fast, model-based structural phylogenetics beyond the twilight zone. bioRxiv, 571181. December 12, 2023. https://www.biorxiv.org/content/10.1101/2023.12.12.571181v1 https://doi.org/10.1101/2023.12.12.571181
The rest of this story has been told in a thread on Twitter (now “X”, ugh), also posted to Bluesky, but honestly, microblogging platforms are kind of lame now, they mostly require logins to read much. So I am re-posting the thread and graphics below.
Twitter thread on Puente-Lelievre et al. (2023), “Tertiary-interaction characters enable fast, model-based structural phylogenetics beyond the twilight zone.” bioRxiv.
A tweetorial explainer on Puente-Lelievre et al (2023). We are very excited about this paper & its implications for phylogenetics. Here’s why! (Work stems from @HFSP grant “How life got moving: reconstructing and re-evolving the bacterial flagellar motor” https://twitter.com/SBSatEd/status/1380517373107666945 )
Protein structure is more conserved than amino acid (AA) sequence. There are many cases where AA identity has decayed towards undetectability, yet the 3D protein structures are superimposable (Illergård et al. 2009) https://onlinelibrary.wiley.com/doi/10.1002/prot.22458 /2

Structural conservation is often exploited in deep homology search and alignment, e.g. in the program Foldseek by @thesteinegger lab. https://www.nature.com/articles/s41587-023-01773-0 . Caroline Puente-Lelievre & I were using Foldseek on deeply-diverged proteins of the bacterial flagellum. /3

We got to thinking - like many before us - about whether we could use protein structure in phylogenetics. Eg Ashar Malik @proteinmechanic (2020) used structural distances & Molecular Dynamics sims to do bootstrapping for uncertainty. Took ~3 weeks. https://academic.oup.com/mbe/article/37/9/2711/5821432 /4
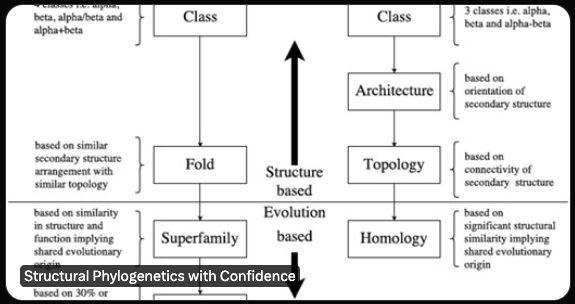
Also, distance-based methods are hard to integrate into modern statistical phylogenetics, which is based on probabilistic models which enable a huge range of tools: Maximum Likelihood & Bayesian inference, statistical model comparison, formal measures of uncertainty, & speed. /5
We played with Foldseek for like a year for homology search, but slowly it dawned on us that its method - converting 3D structures into a 1D sequence of “3Di” states - might be useful not just for rapid structural homology search, but for phylogenetic inference. /6
Then we thought, “Surely someone has thought of this already and therefore it’s either a dumb idea, or it’s coming out next week.” But @AucklandUni has a bunch of structural-phylogenetics-curious, & Caroline, & Jordan Douglas, organized ASPM2023 meeting https://twitter.com/SimonJGreenhill/status/1691570184044339573 /7
All* the cool kids of structural phylogenetics were there: https://twitter.com/phatmattbaker/status/1735052775418183873 (*=not really all, obviously. But, a bunch of people with that interest) /8

At ASPM2023 we realized it was up to us to explore the idea of 3Dis-in-model-based phylogenetics. Moi @steg0s4urus & @thesteinegger lab had just published Foldtree, using 3Di structural alignments to improve AA-distance based phylogenies. https://x.com/thesteinegger/status/1707057683600482813?s=20 /9
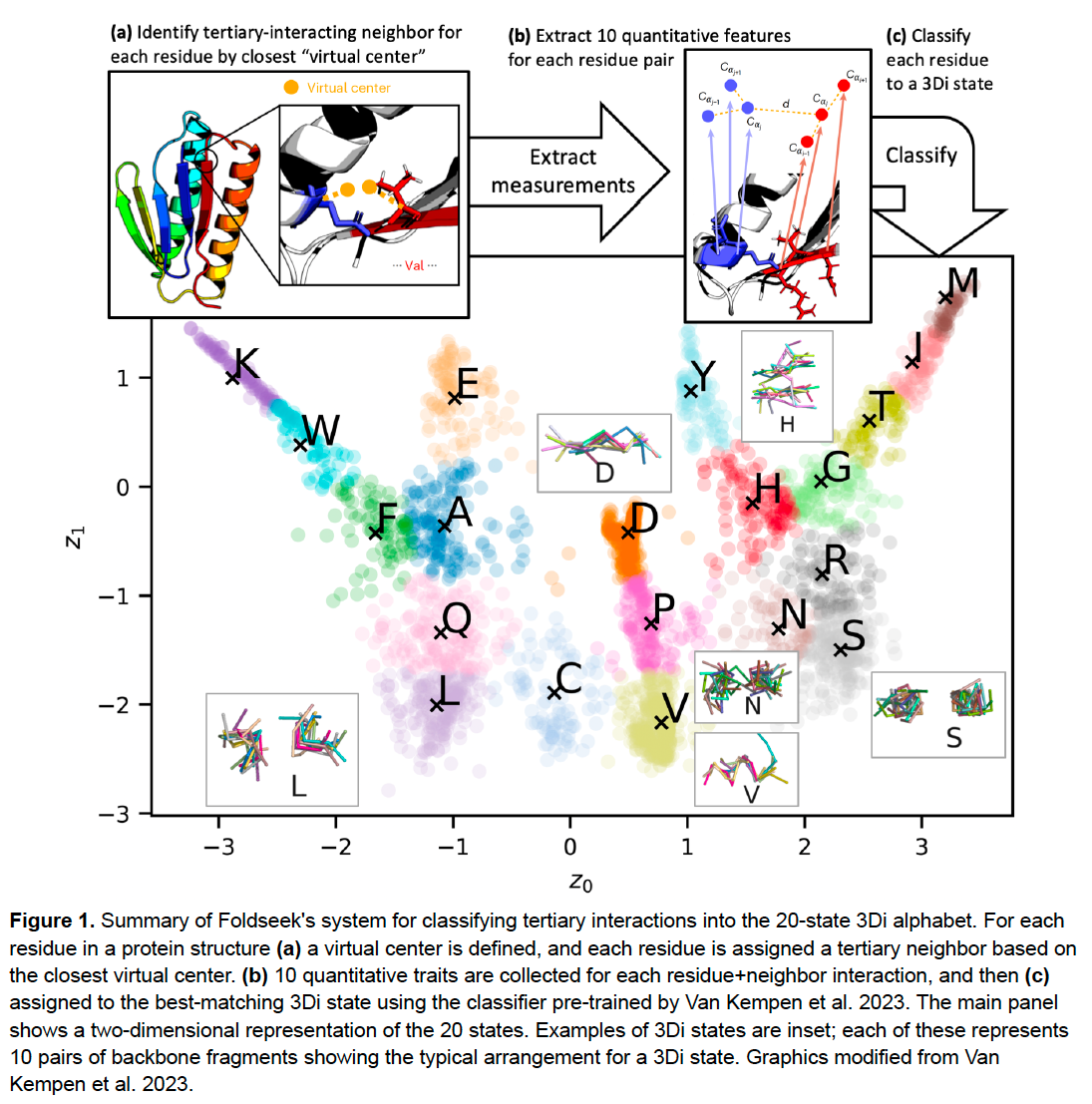
March through the figures of Puente-Lelievre et al. 2023. Here’s a summary of how Foldseek turns a 3D structure file into a sequence of 3Di characters. This takes <1s and gives a 3Di state for each AA in a structure. The Foldseek command is “structureto3didescriptor.” /10
Here is a comparison of AA and 3Di alignments for 53 PDB structures from the Ferritin-like Superfamily, used for “M20”: Malik et al.’s (2020) neighbor-joining distances tree. Coloring is percent identity. Holy moly those 3Di states are CONSERVED! /11
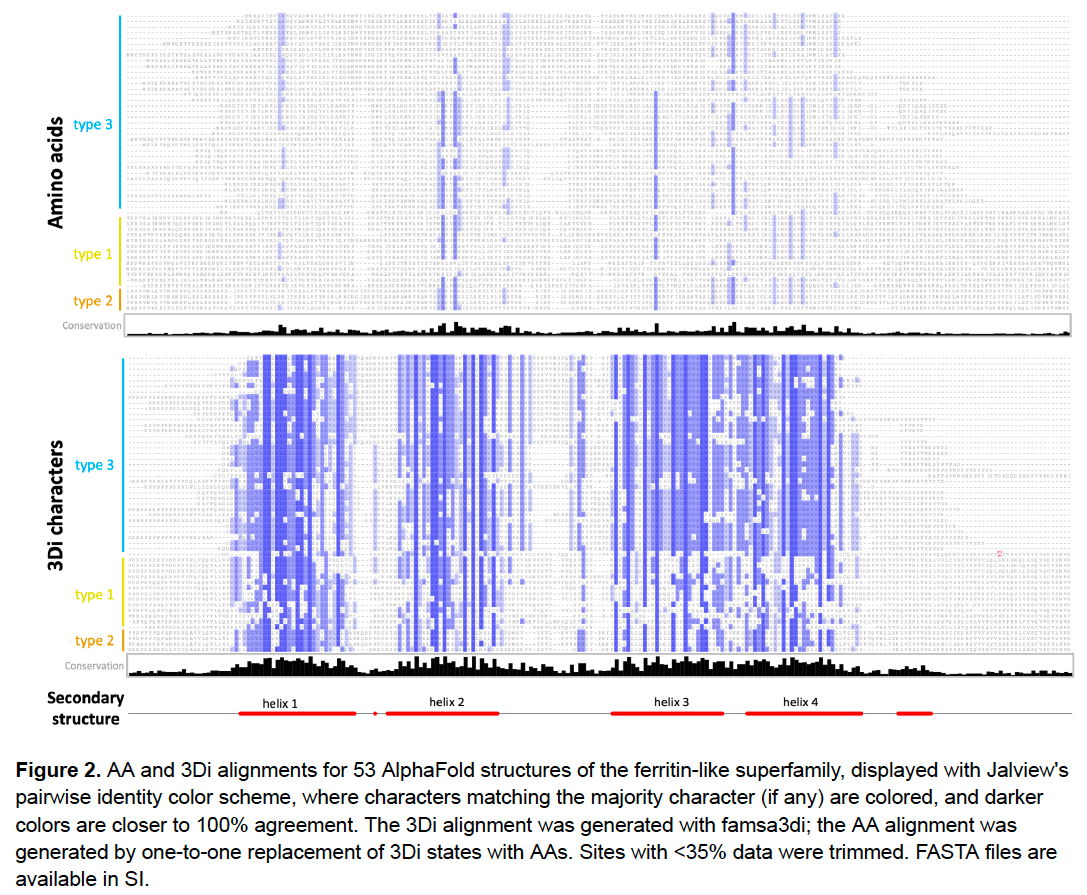
Here’s a zoom-in on part of the alignment. 3Di states seem to have different information than the AAs. If we treat these as phylogenetic characters, would they help resolve things deep in the tree, the “twilight zone” where sequence identity is 30%, 20% or lower? /12

Foldseek has an 3DI cost matrix for alignment calculations. We turned this into a 3DI rate matrix, and tossed it and the AA+3Di characters into the standard Maximum Likelihood phylogenetics program IQtree. Do 3Di characters seem to work like other characters? Yep! /13

We actually ran the data 60 ways in IQtree. The short version: when you use AA+3Di characters, & do all the things you’re supposed to do (structural alignment, model-fitting, partitioning, etc.), you get very close matches to the M20 tree. E.g. with PDB solved structures: /14
Actually, if anything the PDB-derived IQtree analysis is better than M20’s tree: it makes 2 likely groups missed in M20. And: LOOK AT THE RUNTIME. M20’s MD-based bootstrapping took ~3 weeks with parallelization. IQtree took 13.7 min. for 1000 UF bootstraps. On a desktop. /15
If you thought that was cool, look at the next one. This time, we used AlphaFold-predicted structures instead of solved PDB structures. Pretty equivalent tree. But: AlphaFolds are available for…almost everything! Anyone could do this on any dataset! /16 https://alphafold.ebi.ac.uk/
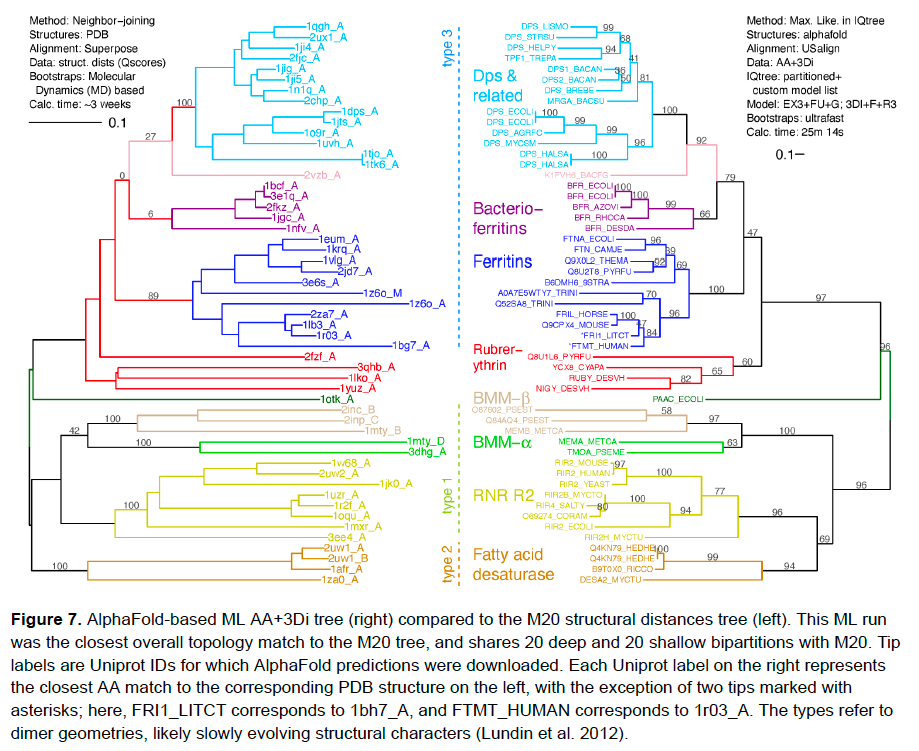
Here’s what we got when we just ran a standard analysis - amino-acids only, and aligned just using amino acids (although with a substitution matrix, MIQS in FAMSA, specialized for divergent proteins). Messy: long-branch attraction artifacts, clades exploding, etc. /17

Summary: this was just a proof-of-concept study, there needs to be study of the many caveats we mention in the manuscript (including philosophical!) etc. But: the future looks bright for structural phylogenetics, and anyone can try it on tough, deep phylogenetics problems! /18
Addendum: Matt Baker at @UNSW aka @phatmattbaker, ringleader of the @HFSP flagellum project, 3rd ranked fencer in Australia in his group, and general amazing guy, has a thread about his path to ASPM2023 and this paper: https://twitter.com/phatmattbaker/status/1735054924512387528 https://twitter.com/phatmattbaker/status/1735052739464585430 /19
Addendum #2: We don’t have a tutorial up yet, but all of the code, commands, example data files etc. are in the Supporting Information zipfile, also available on @github at: https://github.com/nmatzke/3diphy /20
References
Bottaro, A., Inlay, M., and Matzke, N. (2006). “Immunology in the spotlight at the Dover “Intelligent Design’ trial.” Nature Immunology. 7(5), 433-435. May 2006. http://dx.doi.org/10.1038/ni0506-433
Branch, Glenn (2023). “The aftermath of Texas’s disappointing vote on science textbooks.” National Center for Science Education, ncse.ngo. November 27, 2023. https://ncse.ngo/aftermath-texass-disappointing-vote-science-textbooks
Branch, Glenn (2023). “How Mike Johnson Helped Open the Door to Creationism in Louisiana Public Schools.” National Center for Science Education, ncse.ngo. December 11, 2023. https://ncse.ngo/how-mike-johnson-helped-open-door-creationism-louisiana-public-schools
Cain, Trisha Powell (2023). Alabama to update science standards, keep evolution disclaimer. al.com, November 15, 2023. https://www.al.com/educationlab/2023/11/alabama-to-update-science-standards-keep-evolution-disclaimer.html
Foster, James C. (2023). “Scopes Monkey Trial.” Free Speech Center at Middle Tennessee State University. Last updated on September 19, 2023. https://firstamendment.mtsu.edu/article/scopes-monkey-trial/
Illergård, Kristoffer; Ardell, David H.; Elofsson, Arne (2009). “Structure is three to ten times more conserved than sequence – A study of structural response in protein cores.” Proteins, 77(3), 499-508. https://onlinelibrary.wiley.com/doi/10.1002/prot.22458
Matzke, N. (2006). “Design on Trial: How NCSE Helped Win the Kitzmiller Case.” Reports of the National Center for Science Education. 26(1-2), 37-44. Jan-Apr. 2006. https://ncse.ngo/design-trial
Matzke, Nicholas (2015). “The Evolution of Antievolution Policies After Kitzmiller v. Dover.” Science, 351(6268), 10-12. Published online at ScienceExpress on December 17, 2015; print January 1, 2016. http://dx.doi.org/10.1126/science.aad4057 https://www.science.org/doi/full/10.1126/science.aad4057 (Bonus material, media links, FAQ, etc. at: http://phylo.wikidot.com/matzke-2015-science-paper-on-the-evolution-of-antievolution )
Pallen, M., and Matzke, N. (2006). “From The Origin of Species to the Origin of Bacterial Flagella.” Nature Reviews Microbiology. 4(10), 784-790. October 2006. http://www.doi.org/10.1038/nrmicro1493
Puente-Lelievre, Caroline; Malik, Ashar J.; Douglas, Jordan; Ascher, David; Baker, Matthew; Allison, Jane; Poole, Anthony; Lundin, Daniel; Fullmer, Matthew; Bouckert, Remco; Kim, Hyunbin; Steinegger, Martin; Matzke, Nicholas J. (2023). Tertiary-interaction characters enable fast, model-based structural phylogenetics beyond the twilight zone. bioRxiv, 571181. December 12, 2023. https://www.biorxiv.org/content/10.1101/2023.12.12.571181v1 https://doi.org/10.1101/2023.12.12.571181
Scott, E. C., and Matzke, N. (2007). “Biological design in science classrooms.” Proceedings of the National Academy of Sciences. 104(suppl. 1), 8669-8676. May 15, 2007. Part of the v. 104 supplement, “In the Light of Evolution I: Adaptation and Complex Design.” Published online before print May 9, 2007. Also submitted for a Special Volume on the Arthur M. Sackler NAS Colloquium “In the Light of Evolution: Adaptation and Complex Design.” Arnold and Mabel Beckman Center, Irvine, CA, November 30 - December 2, 2006. Organized by John C. Avise and Francisco J. Ayala. http://dx.doi.org/10.1073/pnas.0701505104