
Behe versus ribonuclease; the origin and evolution of protein-protein binding sites
The core concept of Dr. Michael Behe’s recent book “The Edge of Evolution” (Behe, 2007) is that protein-protein binding sites are extremely unlikely to have developed by natural means, and therefore were designed by unknown intelligent agents. There is a lot of interest in this concept, as the tag cloud at PT indicates. A recent paper (Grueninger et al., 2008) on human design of binding sites undermines some of his key assumptions, but what is more interesting is an old paper cited in Grueninger that shows researchers have known for some time that evolution of protein-protein binding sites is not as difficult as Behe makes out. Indeed, his very premise was invalid from the beginning.
Compared to his central image of “Darwin’s Black Box”, the mousetrap, “hard to evolve protein-protein binding” is a far more abstract and much less memorable idea. No matter how deeply misleading the image is a mousetrap at least is something concrete; you can even hold one in your hand. Submicroscopic things that blobbily stick together just don’t catch the imagination in the same way. This may explain why “Edge of Evolution” has had less impact than “Darwin’s Black Box”.
However, to understand the importance of Behe’s claim, consider that many of the proteins in the cell strongly bind to other proteins in order to function. Such protein complexes can range from the simple systems like haemoglobin, the oxygen carrying protein of the body which is made of two separate proteins bound together, to more complex systems like the proteasome, a barrel–shaped complex of 28 subunits which is the garbage bin of the cell that disposes of cellular proteins past their use-by date (Gilles et al., 2003, Valas & Bourne 2008).
Proteins in such complexes generally bind through specific protein-binding sites, which are complementary areas of the two proteins surfaces. Typically a surface loop on one protein (a “bump”) will fit into a corresponding pocket on another protein (a “hole”), so that the proteins fit together like a key fitting into a lock (see figure). Of course, in nature things are rarely this simple. Amino acids can be neutral, negatively charged, positively charged or slightly “oily”. Usually the amino acids of the bump and hole will have complementary charges or “oiliness” as well. On the other hand, the “Lock and Key” image is far too rigid. Proteins are flexible, and will wobble, flex and change shape, so they represent floppy locks and keys, in which a range of shapes may fit.
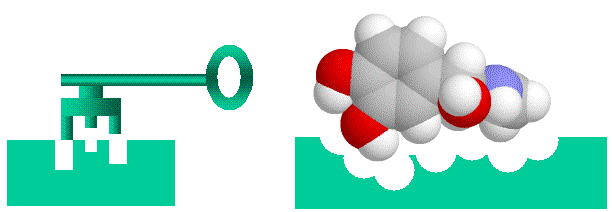 Proteins bind together like a “lock and key” with “bumps” on one molecule matching “holes on the other, electrical charge and oiliness are also factors.
Proteins bind together like a “lock and key” with “bumps” on one molecule matching “holes on the other, electrical charge and oiliness are also factors.
Behe’s basic argument in “EoE” is that protein-protein binding sites contain many interacting amino acids, and that all (or a large majority) of these amino acids must be in place simultaneously for strong, biologically meaningful, protein-protein binding to occur. Note the word simultaneously. As with the “irreducibly complex” mousetrap model, Behe assumes that there is no possible one-mutation-at-a-time path to these binding sites. He doesn’t even mention the possibility in his discussion of binding sites (see the “two-binding site rule”). This is a mirror image version of the Behe & Snoke paper (Behe & Snoke, 2004), that assumed that any binding site could only be reached by multiple neutral mutations, without any role of natural selection. While in the Behe and Snoke paper they calculate the probability of multiple, sequential, neutral mutations in specific locations, in “EoE” Behe calculates the probability of two simultaneous mutations in specific locations.
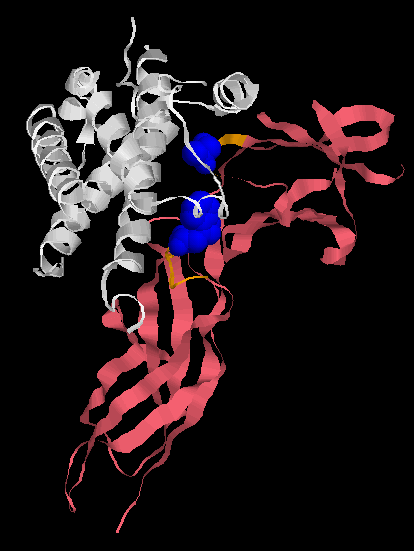 Human Growth Factor receptor (pink) binding to its ligand, Human growth hormone (white). The ligand binding pocket of the Human Growth Factor receptor is shown in orange, but only two amino acids are responsible for the vast majority of the binding (shown in blue). Alternate amino acids can be inserted and high affinity binding still occurs. Unlike Behe implies, you don’t need enormous many amino acids in particular positions to form a binding site.
Human Growth Factor receptor (pink) binding to its ligand, Human growth hormone (white). The ligand binding pocket of the Human Growth Factor receptor is shown in orange, but only two amino acids are responsible for the vast majority of the binding (shown in blue). Alternate amino acids can be inserted and high affinity binding still occurs. Unlike Behe implies, you don’t need enormous many amino acids in particular positions to form a binding site.
Behe claims that for even a simple binding site composed of two amino acids in specific locations that you would need a population of around 1020 organisms to evolve it. Since for large organisms, such as humans, whales, wildebeests and wolverines, this is many orders of magnitude larger than the total population of these organisms over their entire history on this planet, Behe claims we cannot have developed many protein-protein binding sites by natural means. Yet our cells have over 10,000 protein-protein binding sites! Thus, Behe says, multisubunit protein complexes must be the work of a (unknown) designer.
Let’s just stand back for a moment and quickly summarise Behe’s claims:
1) Protein-protein binding sites must be produced by multiple, simultaneous mutations in a specific sequence.
2)There are lots and lots of protein-protein binding sites in modern organisms, far more than could be produced during the lifetime of any given species.
To take the second claim first, note the slight of hand involved. Humans do have lots of protein-protein binding sites, but we didn’t develop them de novo. The vast majority we inherited from our common ancestor with the chimpanzees. That hominid in turn inherited most of its protein binding sites from its ancestors, and so on. Indeed, the majority of the most impressive protein-protein complexes evolved in single celled organisms over hundreds of millions, if not billions, of years. The populations of these organisms far exceed the measly 1020 that Behe invokes. But reading “EoE” Behe certainly gives the impression that all these protein-protein complexes must have evolved relatively recently, without a deep history. Take the proteasome, it didn’t evolve in some slow reproducing, lumbering multicellular organism, it evolved in bacteria back in the deep Precambrian. For someone who claims that he accepts evolution and natural selection, Behe certainly ignores it when considering protein-protein binding. He also ignores a very important aspect of the evolution of binding sites, exemplified by the proteasome. I’ll amplify this later, but for the moment, hold this thought, Behe ignores known details of protein evolution.
Now, after that roundabout introduction, I’ll return to the first point. Behe implicitly assumes that there is no simple, step by step selectable path to strong protein-protein binding (he also implicitly assumes that there is no step by step path to any multiprotein complex). But is his assumption true?
In the paper I introduced at the beginning of this essay, Grueninger et al. were trying to engineer binding sites into proteins. They were able to produce strong protein-protein binding in many cases with a single mutation. Not only that, the proteins produced multimers of a variety of sizes. Let’s repeat that again, a single mutation produced strong protein-protein binding which resulted in protein complexes. And the researchers hadn’t exhaustively tested all possible mutations and binding sites. Now, these complexes were composed of identical proteins, but this is actually quite important and I will elaborate on this momentarily. But buried away in the references was a paper that was even more illuminating.
This paper was looking at the basis of the binding of bovine seminal ribonuclease. Ribonuclease is an enzyme that, as its name suggests, breaks down ribonucleaic acid. These enzymes are typically monomers, but bovine seminal ribonculease is a modified duplicate of standard ribonuclease which is a dimer. The question that researchers were interested in was which mutations were responsible for binding. At stake was a particular model of how proteins bind to each other. To explain this, I have to briefly diverge into a discussion of protein folding.
When proteins are synthesized in a cell, they have to fold up into their final, three dimensional shapes. In this process, loops on the protein chain fit into pockets in the protein chain. Sound familiar? It’s the same process the produces protein-protein binding. One of the simplest ways for two proteins to bind to each other is if the loop of one binds into the pocket of the other (see the diagram). You can see that it would be very simple to set this up. In the end the researchers found there were multiple ways to get ribonulcease to dimerise. One mutation was all it took. So we have evidence that in nature, single mutations are all it takes to produce important protein-protein complexes. And we have had this evidence for sometime. Why didn’t Behe address this?
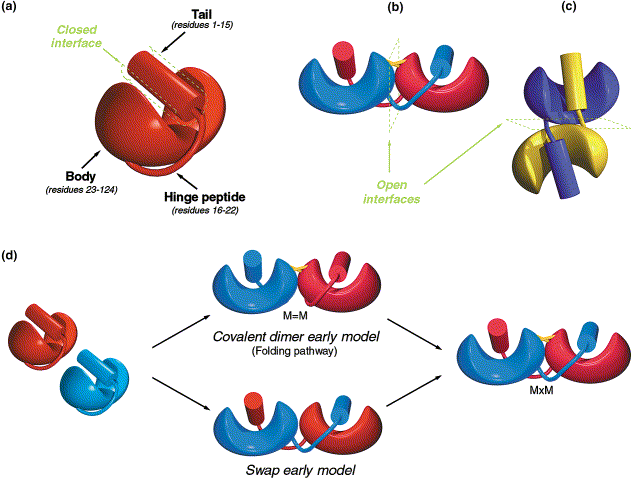 Evolution of homodimer binding, there are two rapid paths to forming homodimers, using internal structures of the protein. Both paths appear to be used, and it takes only a single mutation to generate strong homodimer binding. Diagram taken from Canals et al., 2001.
Evolution of homodimer binding, there are two rapid paths to forming homodimers, using internal structures of the protein. Both paths appear to be used, and it takes only a single mutation to generate strong homodimer binding. Diagram taken from Canals et al., 2001.
We can ask how relevant these results are. Are dimers and multimers of the same protein at all useful? Certainly the bovine seminal ribonuclease is. And indeed, dimers and multimers of the same protein play very important roles. For example, the nicotinic receptor composed solely of five α7 subunits is important in brain function, and there are many similar examples.
As well, these homomultimers are raw material for more complex systems. Duplication and subsequent mutation of the nicotinic a subunit produced beta subunits, in the same way that monomeric haemoglobin became a tetramer of α and β subunits (something Behe accepts). The key issue here is that a mutant α subunit that becomes a β subunit retains the protein-protein binding site, it doesn’t have to reinvent the whole thing again. And the whole process can be repeated several times. There are several crucial nicotinic receptors composed of variant αb complexes. Subsequent duplication and divergence produced the complex αβγδ nicotinic receptor of the skeletal muscle.
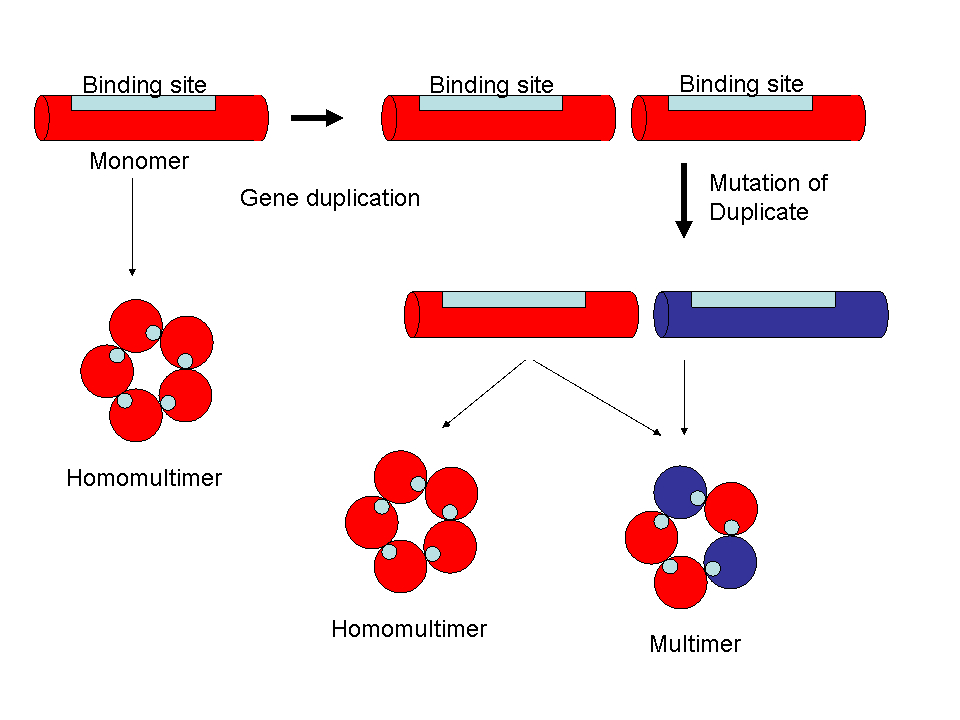 Duplication of the genes for homomultimeric proteins, with subsequent mutation and divergence, can generate large families of interacting proteins without having to generate new binding sites.
Duplication of the genes for homomultimeric proteins, with subsequent mutation and divergence, can generate large families of interacting proteins without having to generate new binding sites.
This process can produce significant complexity, remember the 28 subunit proteasome? Well, in the many primitive organisms, it is constructed solely out a αβ dimer. The dimer is the result of duplication of a simple monomer (and many eubacteria have a simple 12 subunit proteasome composed of a simple monomer which appears ancestral to the dimeric proteasome (Gilles et al, 2003, Valas & Bourne 2008). Subsequent duplication and divergence of the dimer genes produced the 28 subunits that are found in vertebrates, but it all started with a very simple multimer. Again, although Behe says he accepts evolution and natural selection, he ignores the role of evolution in generating structures such as the nicotinic acetylcholine receptor family, the sodium channel family, the potassium channel family, the proteasome, GABA receptors, AAA+ ATPases, NMDA receptors, glycine receptors, histamine H3 receptors … you get the idea.
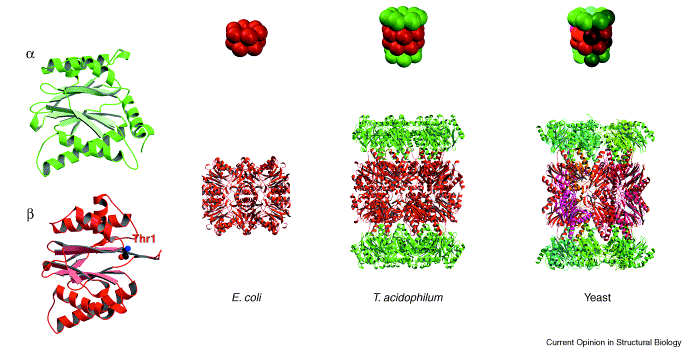 Vertebrate proteasomes are complex barrel like structure of 28 subunits. These evolved by duplication and divergence of a structure made of simple αβ dimers. The α subunit (yellow) itself is a duplicate of the β subunit (red) . The E. coli proteasome is made of a single subunit (red), which shares a common ancestor with the β subunit. Image from Groll et al., 2003
Vertebrate proteasomes are complex barrel like structure of 28 subunits. These evolved by duplication and divergence of a structure made of simple αβ dimers. The α subunit (yellow) itself is a duplicate of the β subunit (red) . The E. coli proteasome is made of a single subunit (red), which shares a common ancestor with the β subunit. Image from Groll et al., 2003
So, a large proportion of protein-protein binding is due to structures that started out as homomultimers, which we have seen can evolve very easily indeed. That’s all very well, but not all protein complexes began as homomultimers. For example, in the voltage-operated calcium channel family each channel is a complex of unrelated proteins. Now, one of Behe’s assumptions is that proteins in protein-protein complexes have no function unless they are in a complex, but in fact this is often not the case. The α subunit of the voltage operated calcium channel is in fact a fully functioning ion channel. The α subunit complexed with either the β subunit alone, or the β and γ subunits modify the characteristics of ion flow through the channel. Again, multiple forms of voltage gated calcium channels are formed by duplication and divergence of the subunits. Again, there is no need to develop entirely new binding sites, these are carried over with the duplicated proteins, and again there are multiple classes of proteins that follow this pattern. For example the NADPH oxidase family are diverged duplicates, which exists as simpler systems in simpler organisms. Even in these kinds of heteromultimeric complexes, some of the elements will be internal duplicates as well, making things simpler.
The scope of this inheritance of binding sites can be illustrated with the G-protein coupled receptor family. The receptor protein is in a complex with the eponymous G-protein [1], through which it signals. The repertoire of vertebrate G-protein coupled receptors are modified duplicates of the original G-protein coupled receptor in single celled organisms. How big is that repertoire? In vertebrates there are around 500 G-protein coupled receptors, that represents a big chunk of the approximately 10,000 protein-protein binding sites that Behe cites that don’t have to be evolved from scratch. Now consider that most protein complexes are parts of families, even before we consider the huge families of protein complexes that evolved from homomultimers, and Behe’s big numbers begin to melt away like snow at Arakarula[2].
We have up to now considering simple point mutations. But there is another way proteins can gain binding sites. That is through gene fusion or crossover, where segments of genes containing binding sites can be swapped into other genes. Indeed, in the protein kinases, new targeting binding sites have been produced in just this way.
Duplication, divergence, crossover, fusion and inheritance; once again, Behe is unacquainted with the evolutionary history of the systems he claims to describe.
However, while there is lots of experimental evidence that single mutations can easily produce high affinity homodimers and homomultimers (which then, by duplication and divergence, result in the protein complexes that we see today), can such simple, one amino acid mutations produce binding between entirely different proteins (say the α and β subunits of the voltage gated calcium channels)? The answer is yes. Once again, we turn to bovine seminal ribonuclease. The simple mutation that made it dimerise has also lead it to bind to a variety of other, unrelated proteins (without any harm to cows). This emphasises the messy, contingent nature of biology, and also illustrates that protein-protein binding is a lot easier to evolve than Behe claims.
Summary
Behe claims that there are a huge number of protein-protein binding sites, and that even one protein-protein binding site is extraordinarily difficult to evolve. However Behe greatly overestimates the difficulty of developing a binding site, ignores the fact that the majority of 10,000 binding sites in modern vertebrates are duplicate copies of each other, with there being only a much smaller number of basic binding motifs and ignores the fact that most of these basic binding motifs were developed in rapidly dividing single celled organisms with very large populations.
Far from protein-protein binding pointing to an unknown designer, protein binding sites point directly to descent with modification and the “tinkering” of natural selection.
[1] Strictly speaking, G-proteins bind to the G-protein-coupled receptors only when the receptors are activated, rather than permanently, as in voltage gated calcium channels. However, G-protein receptor binding involves the same issues of selective interaction of “bumps” and “holes” as with all other protein complexes.
[2] Arkarula in the Flinders Ranges is on the edge of the great Australian Desert, and boy, is it hot!
- Behe M. “The Edge of Evolution: The Search for the Limits of Darwinism”. Free Press, New York. 2007, pp. 135-147.
- Behe MJ, Snoke DW. Simulating evolution by gene duplication of protein features that require multiple amino acid residues. Protein Sci. 2004 Oct;13(10):2651-64. Epub 2004 Aug 31.
- Gille C, Goede A, Schlöetelburg C, Preissner R, Kloetzel PM, Göbel UB, Frömmel C.A comprehensive view on proteasomal sequences: implications for the evolution of the proteasome. J Mol Biol. 2003 Mar 7;326(5):1437-48.
- Groll M, Clausen T. Molecular shredders: how proteasomes fulfill their role. Curr Opin Struct Biol. 2003 Dec;13(6):665-73.
- Grueninger D, Treiber N, Ziegler MO, Koetter JW, Schulze MS, Schulz GE. Designed protein-protein association. Science. 2008; 319:206-209.
- Canals A, Pous J, Guasch A, Benito A, Ribó M, Vilanova M, Coll M. The structure of an engineered domain-swapped ribonuclease dimer and its implications for the evolution of proteins toward oligomerization. Structure. 2001; 9:967-976
- Ciglic MI, Jackson PJ, Raillard SA, Haugg M, Jermann TM, Opitz JG, Trabesinger-Rüf N, Benner SA. Origin of dimeric structure in the ribonuclease superfamily. Biochemistry. 1998; 37:4008-4022.
- Valas RE, Bourne PE.Rethinking Proteasome Evolution: Two Novel Bacterial Proteasomes. J Mol Evol. 2008 Apr 4; [Epub ahead of print]