
Mapping fitness: protein display, fitness, and Seattle
![]() A couple of months ago we started looking at the concept of fitness landscapes and at some new papers that have significantly expanded our knowledge of the maps of these hypothetical spaces. Recall that a fitness landscape, basically speaking, is a representation of the relative fitness of a biological entity, mapped with respect to some measure of genetic change or diversity. The entity in question could be a protein or an organism or a population, mapped onto specific genetic sequences (a DNA or protein sequence) or onto genetic makeup of whole organisms. The purpose of the map is to depict the effects of genetic variation on fitness.
A couple of months ago we started looking at the concept of fitness landscapes and at some new papers that have significantly expanded our knowledge of the maps of these hypothetical spaces. Recall that a fitness landscape, basically speaking, is a representation of the relative fitness of a biological entity, mapped with respect to some measure of genetic change or diversity. The entity in question could be a protein or an organism or a population, mapped onto specific genetic sequences (a DNA or protein sequence) or onto genetic makeup of whole organisms. The purpose of the map is to depict the effects of genetic variation on fitness.
Suppose we want to examine the fitness landscape represented by the structure of a single protein. Our map would show the fitness of the protein (its function, measured somehow) and how fitness is affected by variations in the structure of the protein (its sequence, varied somehow). It's hard enough to explain or read such a map. Even more daunting is the task of creating a detailed map of such a widely-varying space. Two particular sets of challenges come to mind.
1. To make any kind of map at all, we need to match the identity of each of the variants with its function.
2. To create a detailed map, we need to examine many thousands -- or millions -- of variants. This means we need to be able to make thousands of variants of the protein.
So let's take the second challenge first: how do we make a zillion variants of a protein? Well, we can introduce mutations, randomly, into the gene sequence for the protein and use huge collections of those random variants in our analysis. The collection is called a library, and believe it or not, the creation of the library isn't our biggest challenge. Because if the library only contains gene sequences, then it's no use in an experiment on protein fitness. We need our library of gene sequences to be translated into a library of proteins. How are we going to do that? And remember the first challenge: we need to be able to identify each variant. So even if we can get our gene sequences made into protein, how will we be able to identify the sequences after we've mapped the fitness of all the variants?
Or, in simpler terms, here's the problem. It's pretty straightforward to make a library of DNA sequences. And it's pretty straightforward to study the function of a protein. (Note to hard-working molecular biologists and protein biochemists: no, I'm not saying it's easy.) The problem is getting the two together so that we can study the function of the proteins with biochemistry but then identify the interesting variants using the powerful tools of molecular biology. What we need is a bridge between the two.
The bridge most commonly used in such experiments is a technique called protein display. There are a few different ways to do it, but the basic idea is that the DNA sequence is encapsulated so that it remains linked to the protein it creates. One cool way to do this is to hijack a virus and force it to make itself using your library. The virus will use a DNA sequence from your library, dutifully make the protein that is encoded by that DNA sequence, and displaying that protein on its surface. There's our bridge: a virus, with the protein on the surface ready for analysis and the DNA sequence stored inside the same virus. Brilliant, don't you think?
Yes, but there's one more problem to be solved. We said we want to do this millions of times. That means we have to grab the viruses of interest, get the DNA out of them, and read off the sequence of that DNA. (That's how we can identify the nature of the variation.) Millions of times. Methods of protein display provided the bridge, but until very recently a crippling bottleneck remained: the sequencing of the DNA was too time-consuming to allow the identification of more than a few thousand variants at a time.
That was then. This is now: the era of next-generation sequencing, in which DNA sequences can be read at blinding speed and at moderate cost. (A currently popular technology is Illumina sequencing.) These techniques have given us unprecedented capacity to decode entire genomes and to assess genetic variation on genome-wide scales. A few months ago, the same methods were used to eliminate that last bottleneck in the use of protein display, demonstrating how a protein fitness map can be generated simply and at very high resolution. The article is "High-resolution mapping of protein sequence-function relationships" (doi) from Nature Methods, by Douglas Fowler and colleagues in Stan Fields' lab at the University of Washington.
The experiment focused on one interesting segment of one protein. The segment is called a WW domain and it's an interesting building block which is found in various proteins and which mediates interactions between different proteins. (A sort of docking site.) The authors chose the WW domain both for its interesting functions and because it has been used in protein display experiments of the type they performed. Then they created their tools.
1) They generated a library of more than 600,000 variants of the domain, displayed on the surface of their chosen bridge -- the T7 bacteriophage (a virus that targets bacteria).
2) They designed a means to assess the function of the variants. Because the function of the WW domain is docking, they used docking as their functional criterion, and then devised a straightforward system to detect the strength of the binding of the variants to a typical docking partner. (For the biochemically inclined, they used a simple peptide affinity binding assay on beads.)
Then the key experimental step: the authors used that system to select the variants that can still bind. In other words, they selected the functional variants. The selection step was moderate in strength, and the idea is that variants that bind really well will be enriched at the expense of variants that bind less well. Variants that don't bind at all will be removed from the library.
They repeated the selection step six times in succession. So, the original library was subjected to selection, generating a new library, which was subjected to selection again, and so on, until the experimenters had six new libraries. Why the repetition? It's one of the really smart aspects of the experiment and it has to do with the strength of selection. If selection were quite strong, such that only the strongest-binding variants survive, then the analysis will just yield a few strong-binding variants. That's a simple yes-or-no question, providing no information about the spectrum of binding that can be exhibited by the variants. Instead, the authors tuned the system so that selection is moderate, leading to enrichment but not complete dominance of the stronger-binding variants. Recall that binding represents fitness in this experiment; this means that the authors subjected their population to a moderate level of selection in order to map the fitness of a large number of variants. By repeating the selection, they could watch as some variants gradually increased in frequency. Sounds kind of like evolution, doesn't it?
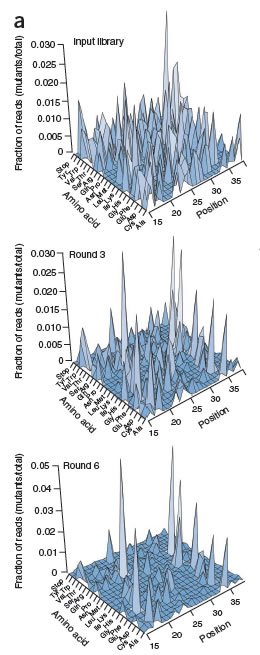
Finally, the scientists subjected those libraries to Illumina sequencing, thus closing the loop between function and sequence. (In genetic terms, we would say that they closed the loop between phenotype and genotype.) And at that point they were able to draw fitness landscapes of unprecedented resolution, shown in the graphs on the right. The top graph shows the original library. The height of each peak represents frequency in the library, and the two horizontal axes represent each possible sequence of that WW domain. Notice that the original library is complex and diverse, as indicated by numerous peaks on the graph. The second and third graphs show the library after three and six rounds of selection. Note the change in the number of peaks and in their relative sizes: selection has reduced the complexity of the library, removing variants that are far less fit and altering the relative amounts of the survivors. The first three rounds of selection reduced the library to 1/4 the original size, and after six rounds it was down to 1/6 original size, but still contained almost 100,000 variants.
The bottom graph, then, is a fitness landscape, of a segment of a protein, at very high resolution. More technically, it depicts the raw data (relative amounts of surviving variants) that the authors used to determine relative fitness; to make that assessment, they calculated "enrichment ratios" to account for the fact that the initial library didn't contain equal amounts of each variant. These enrichment data enabled them to calculate the extent to which each point in the sequence is amenable to change, and then to identify the particular changes at those points that led to changes in fitness. Now that's high resolution.
The power of approaches like this should be obvious: disease-related mutations can be identified in candidate genes, and the same approach can be used to map the landscape of resistance to drugs in pathogens or cancer cells. And, of course, evolutionary questions of various kinds are much more tractable when tackled with methods like this. The authors expect the payoff to be immediate:
Because the key ingredients for this approach -- protein display, low-intensity selection and highly accurate, high througput sequencing -- are simple and are becoming widely available, this approach is readily applicable to many in vitro and in vivo questions in which the activity of a protein is known and can be quantitatively assessed.
Now, given these vast opportunities now available to scientists interested in protein evolution, wouldn't you think that design theorists who write on the topic will be eager to get involved in such studies? I sure would, especially since the lab that did this work is within a short drive of the epicenter of intelligent design research, a research insitute headed by a scientist whose professional expertise and interest lies in the analysis of protein sequence-function relationships. As I've repeated throughout this series, there's something strange about a bunch of scientists who want to change the world but who can't be bothered to interact with the rest of the scientific community, a community that in this case is well-represented in active laboratories right down the road. (I'm eager to be proven wrong on this point, by learning that ID scientists have interacted with the Loeb lab or the Fields lab.)
More to the point, there's something tragically ironic about the fact that the ID movement is headquartered in Seattle, inveighing against "Darwinism" while obliviously amidst a world-class gathering of scientists who are busy tackling the very questions that ID claims to value.
(Cross-posted at Quintessence of Dust.)
-------
Fowler, D., Araya, C., Fleishman, S., Kellogg, E., Stephany, J., Baker, D., & Fields, S. (2010). High-resolution mapping of protein sequence-function relationships Nature Methods, 7 (9), 741-746 DOI: 10.1038/nmeth.1492