
Meyer's Hopeless Monster, Part III
The paperback edition of Stephen Meyer’s book Darwin’s Doubt: The Explosive Origin of Animal Life and the Case for Intelligent Design has just been published. It has a new chapter responding to critics of the book – Donald Prothero, Charles Marshall, and yours truly, the blogger the ID guys were dismissing for a year based on the fact that I wrote the review quickly. The largest section of the new chapter responds to me.
The response shows Meyer is finally improving on a few issues like crown/stem group thinking, but rather like a student who flunked the midterm of a phylogenetics course and decided to finally start paying attention, Meyer still makes huge, amateur mistakes. I’ll highlight a few.
(Unfortunately, I have no time to do a detailed rebuttal, so this will just be quick notes. I am frantically trying to finish projects before meeting season starts in, hoo boy, two days. First I am going to SMBE 2014 in Puerto Rico to present this work, and then to Evolution 2014 in Raleigh, NC to present this work.
(By the way, see part of my presentation, cool animations of stochastic mapping of possible biogeographic histories under different models, below…)
Caption: Stochastic mapping of approximately equiprobable alternative histories under each model. Left: DEC model. Right: DEC+J, which includes founder-event speciation. Key: Blue, K=Kauai. Green: O=Oahu. Yellow: M=Maui-Nui. Red: H=Hawaii Big Island. Kauai is the oldest high island (~5.2 Ma), the Big Island is the youngest island (~0.5 Ma).

.
Background – previous posts on the Cambrian/Meyer
Alan Gishlick, Nick Matzke, and Wesley R. Elsberry (2004). “Meyer’s Hopeless Monster.” Panda’s Thumb post, August 24, 2004. http://pandasthumb.org/archives/2004/08/meyers-hopeless-1.html
The “Meyer 2004” Medley - The Panda’s Thumb – the complete history of the Meyer 2004 craziness.
Matzke, Nicholas (2005). Down with phyla! - The Panda’s Thumb, which reviewed:
- David Fitch and Walter Sudhaus, “One small step for worms, one giant leap for ‘Bauplan?’” Evolution & Development 4:4, 243-246.
- Budd, G. E. and S. E. Jensen. 2000. “A critical reappraisal of the fossil record of the bilaterian phyla. Biological Reviews of the Cambridge Philosophical Society 75:253-295.
- Graham Budd (2001). “Climbing life’s tree.” Nature 412, 487.
- Jaume BaguÃÂñÃÂàAnd Jordi Garcia-FernÃÂàndez (2003). “Evo-Devo: the Long and Winding Road.” Int. J. Dev. Biol. 47: 705-713. PubMed
- Walter Sudhaus (2004). “Radiation within the framework of evolutionary ecology.” Organisms, Diversity & Evolution 4, 127-134.
- Gonzalo Giribet (2003). “Molecules, development and fossils in the study of metazoan evolution; Articulata versus Ecdysozoa revisited.” 106: 303-326.
Matzke, Nicholas (2005). Down with phyla! (episode II) - The Panda’s Thumb
Matzke, Nicholas (2007). Meet Orthrozanclus (down with phyla!) - The Panda’s Thumb
Matzke, Nicholas (2013). “Meyer’s Hopeless Monster, Part II.” The Panda’s Thumb.
Matzke, Nicholas (2013). “Luskin’s Hopeless Monster.” The Panda’s Thumb.
Matzke, Nicholas (2013). “Meyer on Medved: the blind leading the blind.” The Panda’s Thumb.
.
Statistics and Phylogenetics: the Consistency Index (CI)
Meyer discusses – for the first time ever – the Consistency Index (CI), which is a measure of the congruence of characters on a tree, and a standard statistic calculated in cladistic analysis to assess the treelike nature of the data. Meyer cites two CI values from cladistic analyses of Cambrian groups – 0.565 (Legg et al. 2012) and 0.384 (Briggs and Fortey 1989) and declares them “low”. In the case of Briggs and Fortey (1989), Meyer quotes the authors, who call 0.384 “rather low.” Meyer doesn’t mention that this was just about the very first preliminary attempt at cladistics of Cambrian arthropods, but that’s not the most important problem.
The most important problem is that you can’t just eyeball a CI value for a dataset and decide if it is “high” or “low”. Intuitions based on letter grades (70% is a C, 80% is a B, or whatever) are a poor guide to using a technical statistic. Yes, sometimes scientists themselves eyeball a CI and declare it high or low based on intuition, but only when they have not been sufficiently trained on the topic.
Here’s the reality. This is Figure 1.2.1 of Doug Theobald’s Macroevolution FAQ, derived originally from Figure 6 of Klassen et al. 1991:
Figure 1.2.1. A plot of the CI values of cladograms versus the number of taxa in the cladograms. CI values are on the y-axis; taxa number are on the x-axis. The 95% confidence limits are shown in light turquoise. All points above and to the right of the turquoise region are statistically significant high CI values. Similarly, all points below and to the left of the turquoise region are statistically significant low values of CI. (reproduced from Klassen et al. 1991, Figure 6).
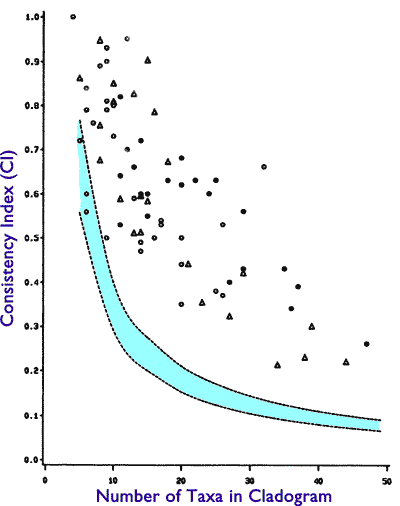
What is the expected CI value if there is no phylogenetic signal in the data? This is what creationists are claiming when they claim the data doesn’t support a phylogenetic tree. This null expectation is easy to calculate (as I mentioned in my original review, but which Meyer, incredibly, missed) by reshuffling each character’s data by randomly assigning the character states to species without regard to phylogeny. The resulting dataset will have the exact same percentages of each character state, the same number of states per character, etc., but will have no phylogenetic signal. Parsimony inference of cladograms can be performed, and CI statistics calculated, for these reshuffled datasets.
The result is a null distribution of CI values. The 95% confidence interval of this null distribution is displayed on the plot. As you can see, the null expectation changes somewhat depending on the number of species in the analysis. So, a CI of 0.5 is low if you have only 10 taxa, but it’s high if you have 30. What is key is that if your dataset’s CI is higher than the null, you can statistically reject the hypothesis of no tree structure in the data. With a little more work you could calculate how many standard deviations you are above the mean of the null distribution.
Briggs & Fortey (1989) had 28 taxa in their analysis. Legg et al. 2012 had 173. Now, consult the figure. I would want to do the randomization myself on the original datasets to be really sure, since conceivably the detailed results could depend e.g. on the number of characters with more than two states, but most morphology datasets are substantially binary characters anyway. As you can see, 28 taxa and a CI of 0.384 is a highly significant rejection of the hypothesis of no cladistic structure, and a CI 0.565 with 173 taxa is an incredibly, mind-bogglingly strong rejection of the null hypothesis. It’s probably hundreds of standard deviations above the random expectation.
Even worse, Meyer should have known about this. Not only has this finding about CI been in the literature since 1991, it’s been prominently available in Theobald’s common ancestry FAQ for 10 years! Meyer himself even cited the FAQ in Darwin’s Doubt, dismissing the entire thing in barely a sentence with “In reality, however, the technical literature tells a different story” (Meyer 2013, p. 122).
The only place where I’ve seen the argument “my gut says that’s a low CI value, therefore cladistics doesn’t support common ancestry” before is from Casey Luskin, Meyer’s “research” assistant for Darwin’s Doubt. Meyer, get a new research assistant! Luskin, get educated before blabbing about technical topics you know nothing about!
Also, regarding Meyer’s argument that a CI of 0.565 means that the 0.435 fraction of the data exhibits homoplasy – the right answer here is “so what”? It is true that the “best characters” are ones that only change state a single time in all of evolutionary history. But character states that evolve twice on a tree still retain a huge amount of phylogenetic signal. Two changes is many fewer changes than you would get if you flipped a coin to place character states at the tips of a tree. Two changes still means that many taxa on the tree share the character state because of common ancestry. If you were going to attempt a classification based on that single character, of course, that wouldn’t work well – but only taxonomists from ancient times, and creationists, think that single-character classifications are how things should work. If you have 100 characters, each of which evolved twice on a tree, the tree would still be readily resolvable.
As usual, creationists make the perfect the enemy of the good. They completely fail understand the difference between “classic homologies” – like the tetrapod limb – and the typical characters used in modern “get as many characters as you can” analyses. In the latter, organisms are atomized as finely as possible while avoiding coding the same character twice under two descriptions. Yes, it would be highly problematic to suggest that the tetrapod limb evolved twice independently. But if you take that limb, and atomize into 100+ individual characters, including the bumps and twists of each bone – is it possible that some of those bumps arose twice independently? Heck yes! If they arose once, they could arise several times, because these highly atomized characters are quite simple (unlike the whole tetrapod limb). Does this homoplasy invalidate the entire enterprise of cladistics and verifying common ancestry? Heck no! Some of those characters might have homoplasy, some might have such high homoplasy that they retain no phylogenetic signal at all (a different thing) – but if you have lots of characters, it doesn’t matter. All that is required is that, on average, most characters retain some phylogenetic signal. Like any measurement of anything in science, you add up many small independent pieces of evidence, some of them noisy or problematic, and yet the final result can be statistically very strong.
This is the typical result of a phylogenetic analysis, and the typical conclusion is that the statistical tree signal is real and quite strong. And this applies to studies of Cambrian taxa (particularly the more recent ones) as much as anywhere else.
Even David Berlinski admitted that the tree structure in cladistic data was real, back in the first round of responses to my review, so I’m not sure where Stephen Meyer thinks he got a leg to stand on on this one. Probably we can just chalk it up to poor research. As usual.
.
Ghost lineages and incongruence between cladograms and stratigraphy
Somehow, in his new chapter, Meyer manages to spend pages on ghost lineages and incongruence between cladograms and stratigraphy, without realizing there is a whole literature on statistically measuring this (despite Meyer’s mentions of “statistical paleontology” in the main text, he has almost no familiarity with the field). The typical result of such tests is that there is overall congruence between the two, again a statistically strong pattern.
Again, Theobald covered it thoroughly, and Meyer and Luskin just missed it. I think their brains must just short circuit when they see Theobald’s FAQ:
The most scientifically rigorous method of confirming this prediction is to demonstrate a positive corellation between phylogeny and stratigraphy, i.e. a positive corellation between the order of taxa in a phylogenetic tree and the geological order in which those taxa first appear and last appear (whether for living or extinct intermediates). For instance, within the error inherent in the fossil record, prokaryotes should appear first, followed by simple multicellular animals like sponges and starfish, then lampreys, fish, amphibians, reptiles, mammals, etc., as shown in Figure 1. Contrary to the erroneous (and unreferenced) opinions of some anti-evolutionists (e.g. Wise 1994, p. 225-226), studies from the past ten years addressing this very issue have confirmed that there is indeed a positive corellation between phylogeny and stratigraphy, with statistical significance (Benton 1998; Benton and Hitchin 1996; Benton and Hitchin 1997; Benton et al. 1999; Benton et al. 2000; Benton and Storrs 1994; Clyde and Fisher 1997; Hitchin and Benton 1997; Huelsenbeck 1994; Norell and Novacek 1992a; Norell and Novacek 1992b; Wills 1999). Using three different measures of phylogeny-stratigraphy correlation [the RCI, GER, and SCI (Ghosts 2.4 software, Wills 1999)], a high positive correlation was found between the standard phylogenetic tree portrayed in Figure 1 and the stratigraphic range of the same taxa, with very high statistical significance (P [LESS THAN] 0.0001) (this work, Ghosts input file available upon request).
As another specific example, an early analysis published in Science by Mark Norell and Michael Novacek (Norell and Novacek 1992b) examined 24 different taxa of vertebrates (teleosts, amniotes, reptiles, synapsids, diapsids, lepidosaurs, squamates, two orders of dinosaurs, two orders of hadrosaurs, pachycephalosaurs, higher mammals, primates, rodents, ungulates, artiodactyls, ruminants, elephantiformes, brontotheres, tapiroids, chalicotheres, Chalicotheriinae, and equids). For each taxa, the phylogenetic position of known fossils was compared with the stratigraphic position of the same fossils. A positive correlation was found for all of the 24 taxa, 18 of which were statistically significant.
As a third example, Michael Benton and Rebecca Hitchin published a more recent, greatly expanded, and detailed stratigraphic analysis of 384 published cladograms of various multicellular organisms (Benton and Hitchin 1997). Using three measures of congruence between the fossil record and phylogeny (the RCI, SRC, and SCI), these researchers observed values “skewed so far from a normal distribution [i.e. randomness] that they provide evidence for strong congruence of the two datasets [fossils and cladograms].” Furthermore, Benton and Hitchin’s analysis was extremely conservative, since they made no effort to exclude cladograms with poor resolution, to exclude cladograms with very small numbers of taxa, or to use only fossils with reliable dates. Including these types of data will add confounding random elements to the analysis and will decrease the apparent concordance between stratigraphy and cladograms. Even so, the results were overall extremely statistically significant (P [LESS THAN] 0.0005). As the authors comment in their discussion:
“… the RCI and SCI metrics showed impressive left-skewing; the majority of cladograms tested show good congruence between cladistic and stratigraphic information. Cladists and stratigraphers may breathe easy: the cladistic method appears, on the whole, to be finding phylogenies that may be close to the true phylogeny of life, and the sequence of fossils in the rocks is not misleading. … it would be hard to explain why the independent evidence of the stratigraphic occurrence of fossils and the patterns of cladograms should show such striking levels of congruence if the fossil record and the cladistic method were hopelessly misleading.” (Benton and Hitchin 1997, p. 889)
Additionally, if the correlation between phylogeny and stratigraphy is due to common descent, we would expect the correlation to improve over longer geological time frames (since the relative error associated with the fossil record decreases). This is in fact observed (Benton et al. 1999). We also would expect the correlation to improve, not to get worse, as more fossils are discovered, and this has also been observed (Benton and Storrs 1994).
As for Cambrian taxa, I don’t believe this sort of analysis has been done yet (I could do it – if you put me on your grant/paper!). But the available data already indicates the basic result will be the same as the above. The oft-mentioned pattern of “we see mostly stem groups in the early Cambrian” lines up extremely well with the stratigraphy-cladogram congruence pattern discovered elsewhere.
.
Other problems with Meyer’s new chapter, briefly mentioned
-
Meyer repeats the usual creationist silliness about transitional fossils, claiming that nothing except direct ancestors count, and cladistics can’t provide these, so it’s all meaningless. He even adds a quote from someone with old-school pattern cladist tendencies, in this case Henry Gee. Par for the course, and it completely ignores what the realistic expectations are when you take a phylogeny and sample it with just a few lagerstatten over 10s of millions of years, each of them sampling one narrow time period at one location. This isn’t the late Cenozoic, where we can get pretty continuous and geographically representative samples of some lineages, enough to say that, say, Homo erectus is very likely the species ancestral to Homo sapiens. It’s utterly ridiculous to expect that kind of precision in the Cambrian. Thus we look for familial relationships and collateral ancestry. The methods for determining these sister-group relationships are rigorous and well-tested, as discussed in my original review and in Theobald’s FAQ, all of it completely ignored in Meyer’s response.
-
Meyer acts surprised about the term “collateral” ancestry, despite prominent discussions in, say, Prothero’s Evolution: What the Fossils Say and Why it Matters (p. 82), Padian’s testimony in Kitzmiller, or, say, Darwin (1859).
-
Meyer repeats his statements about how cladistics doesn’t show how new information and developmental changes come about – No, cladistics shows the major steps that occurred and their order, and that disproves the idea that it had to happen all at once in defiance of Darwinian gradualism, which is a key feature of Meyer’s argument. Once you have the basic steps and their order, then evo-devo and other disciplines can work on each of the steps. Meyer’s tactic of requiring field A to answer question Z’ and field Z to answer question A’, and never putting together the amazing idea that field A might answer A’ and field Z might answer Z’, is widespread with other creationists (e.g. Paul Nelson). It’s a neat trick, but it will only work on people who are uninformed about these fields.
-
Meyer again ignores the voluminous, detailed, published work on the evolution of new genes, with a vague hand wave about how all that biologists can show is merely “reshuffling information.” I addressed the wild unreality of creationist failures to deal with the work and data in the “evolutionary origin of new genes” subfield in my original review. Meyer just ignores this. Again, even David Berlinski and Michael Behe have admitted that normal evolutionary processes can produce new genes. What’s Meyer’s problem? Where’s the line, Stephen Meyer? And don’t say “protein domains”, because you still haven’t shown that new protein domains had much of anything to do with the Cambrian Explosion. Most protein domains go way back to single-celled eukaryotes and to prokaryotes.
-
Meyer mentions small shellies briefly – mostly referencing his previous half-baked response online (summary paraphrase: “I briefly mentioned them in passing in a footnote, so I’m good, and Marshall’s diagram draws their diversity separately from the Cambrian phyla, so they must not be connected, so I was right to decide to ignore them, even though it was probably a straight-up mistake rather than a decision.”). In the new chapter, it is apparent that in Meyer’s head, the small shellies are a totally separate event from the Cambrian explosion, as if the gradually increasing diversity and complexity in fossil shells in the 15 million years just before the classic “explosion” was just mere coincidence. It also ignores that some of the small shellies have been taxonomically connected to classic phyla, e.g. when a rare fossil is found showing how the small pieces of “chain mail” body armor assemble together to cover a larger animal. The apparently piecewise evolution of skeletons is, I suppose, also just another coincidence for Meyer.
.
Conclusion
A fair bit more could be said, but, as before, this is enough to show that Meyer is not a serious scholar on this issue. He’s still making student-level mistakes.
I am interested in further discussion in the comments, as long as it doesn’t get nasty.